J Pharm Pharmaceut Sci (www.cspscanada.org) 10(1):26-36, 2007
The metabolism of midazolam and comparison with other CYP enzyme substrates during intestinal absorption: in vitro studies with rat everted gut sacs
Cécile Arellano, Claude Philibert, Christelle Vachoux, John Woodley, Georges Houin
Laboratoire de Cinétique des Xénobiotiques, Faculté des Sciences Pharmaceutiques, Université Paul Sabatier Toulouse III, 31062 Toulouse cedex 9, France. Laboratoire de Pharmacocinétique et Toxicologie Clinique, Institut Fédératif de Biologie, Hôpital Purpan, 330 avenue de Grande Bretagne, 31059 Toulouse cedex 9, France.
Received, October 13, 2006; Revised February March 3, 2007; Accepted, March 6, 2007,Published March 6th 2007
Corresponding Author: Dr. Cécile Arellano, Tel / FAX: +33 (0)5 62 25 68 85 E-Mail: arellano@cict.fr
ABSTRACT - PURPOSE. The purpose of this study was to quantify the intestinal metabolism of midazolam, a CYP P450 substrate, usually used as a probe for the activity of the isoform CYP3A4/1 and to compare it with previous results obtained for other P450 substrates such as testosterone, dextromethorphan and bupropion, which show some specificities for different CYP isoforms. The aim was to shed light on the role of metabolism in the intestinal tissues and the relationship with efflux mechanisms, such as by P-glycoprotein (P-gp) and the influence of metabolism on bioavailability. METHODS. We used the improved everted rat gut sac model to study in vitro the absorption and metabolism of the different CYP isoenzyme probes: midazolam, testosterone, bupropion and dextromethorphan. This method enables drug metabolism to be studied during absorption, conditions which mimick the in vivo situation. The drugs and their metabolites were measured by LC-MS in the mucosal and serosal media and in the mucosal tissue, to give a complete picture of the transport and metabolism. RESULTS. Midazolam, as with the other CYP probes, was metabolized in everted gut sacs. The metabolites were detected in the same proportions in both the serosal and mucosal compartments for midazolam, testosterone and bupropion. In the case of dextromethorphan, the metabolite methoxymorphinan was found at a higher concentration in the mucosal compartment, indicating efflux from the cells. The transport of dextromethorphan and its metabolite was not modified in the presence of verapamil, a P-gp inhibitor, thus demonstrating that dextromethorphan and methoxymorphinan were not P-gp substrates. CONCLUSION. Given that the rat is a widely used species for pre-clinical studies, the everted gut sac model provides a useful tool to assess the role of metabolism during drug absorption by the intestine and is also capable of demonstrating P-glycoprotein mediated transport.
Introduction
The metabolism of drugs when passing through the intestinal mucosa has been the focus of attention in recent years because the intestine plays an important role in the biotransformation of xenobiotics that may undergo first-pass metabolism in the gastrointestinal tract (GIT) (1-4). In addition to hepatic metabolism, high rates of gastrointestinal drug oxidation have been observed in humans and rats. Among the large number of drugs exhibiting low systemic availability (less than 50% oral bioavailability), many are substrates of CYP3A, a major constitutive enzyme of liver and intestine (5-7). Clinical studies of orally administered drugs such as cyclosporin, midazolam, nifedipine and tacrolimus have shown that the oral bioavailability was significantly reduced by intestinal metabolism (8). Different strategies have been used to study the contribution of the intestine to biotransformation and to determine its role relative to hepatic metabolism in the first-pass elimination of drugs. For example, Thummel et al. (9) deduced the proportion of the intestinal metabolism of midazolam from a comparison of midazolam disposition after oral and intravenous administration of the compound to healthy volunteers. They also carried out separate in vitro studies with human intestinal and hepatic microsomes and concluded that the first-pass extraction ratios of midazolam by the gut and liver were comparable. An intestinal multilumen perfusion technique was proposed to assess directly the absorption, metabolism and transport of verapamil in the human intestinal wall (10) and a single pass intestinal perfusion model was recently proposed in rats to study the modulation of intestinal metabolism by the efflux transporter P-glycoprotein (P-gp) (11). Diltiazem metabolism and the efflux of desacetyl-diltiazem by P-gp in rat jejunum have been demonstrated using an in vitro chamber system (12).
In vitro models are more often used than in vivo investigations to study gut metabolism and intestinal microsomes from experimental animals have been prepared by various methods in different laboratories (13-16). However, the results vary and appear to be very dependant on the procedure used to prepare the microsomal fractions (17). Caco-2 and other intestinal cells in culture have also been used to study intestinal metabolism and inhibitory or induction related drug interactions, but normal Caco-2 cells have a very low expression of the metabolising enzymes (18-20).
Although they may play separate roles in restricting absorption, it has been proposed that P-gp and CYP3A4 act synergistically to increase pre-systemic drug metabolism, partly because of the extensive overlap in their substrate specificity (21-24). As has been reviewed recently (25), different attempts had been made to find an appropriate in vitro model expressing both CYP3A4 and P-gp. For example, polarized cells co-expressing CYP3A4 and MDR1/P-glycoprotein (P-gp) have been especially developed to study the interplay between drug transport and drug metabolism at the level of CYP regulation. The different approaches used to develop these cellular model systems with LLC-PK1 or MDR1 cells, stably expressing P-gp, and transfected with different CYP3A vectors (26) unfortunately give a wide variability in CYP 3A expression. The metabolism of indinavir has been studied with vitamin D3-induced Caco-2 cells, in conditions where Caco-2 cells co-express CYP3A4 and P-gp (27). The results obtained supported a role for P-gp in increasing intestinal pre-systemic CYP3A-generated metabolites from the intracellular compartment, but, under the same conditions, the secretion of 6--hydroxytestosterone across the apical membrane was not observed, showing that not all metabolites produced by CYP3A4 are effluxed by P-gp. P-gp also appeared to increase the intestinal metabolism of indinavir after its oral administration to rats pre-treated with dexamethasone to induce the metabolic enzymes (28).
The problem with these methods is that they give no information on the actual activities of the enzymes in the epithelial cells and the effect this may have on the first-pass metabolism of a drug during its absorption by the intestine. As concluded by Hochman and al. (27), an in vitro model capable of emulating the transport properties of the intestinal epithelium that expresses both CYP3A4 and P-gp would provide a useful tool for the assessment of the contribution of both activities. With this latter objective in mind, we have turned to the use of our improved everted gut sac system which has proved very valuable for studying various aspects of drug absorption in vitro (29-31) including P-gp activity (32) and intestinal metabolism (33). The novelty of this system is the ability to study drug metabolism during the process of absorption. In order to identify which CYP enzymes are actually active during drug absorption, we have developed sensitive LC-MS methods to analyse the following CYP specific substrates and their metabolites: testosterone (34), dextromethorphan (35), bupropion (36).
We now report results with midazolam which, like testosterone, was a specific probe used for the metabolic activity of the P450 CYP3A4/1 enzymes. Additional experiments have been carried out with the previously studied drugs: testosterone, dextromethorphan and bupropion to emphasize the usefulness of the everted gut sac model in the evaluation of intestinal absorption. We have now related the interaction of these drugs with gut mucosa to their lipophilicity and quantified the amount of compounds accumulated in gut tissues and the quantity of metabolites found on both sides of the gut mucosa. The effect of verapamil on dextromethorphan uptake and metabolism has also been examined to assess the potential role of P-gp on its absorption and metabolism.
Materials and METHODS
Chemicals
Midazolam, hydroxymidazolam and bromazepam were a kind gift from Roche Laboratories (Neuilly-sur-Seine. The origin of testosterone, dextromethorphan, bupropion and related metabolites has been detailed in our previous reports (34-36). Ultrapure water was obtained using a Millipore Simplicity 185 apparatus. Dichloromethane, hexane, diethylether, and methanol (SDS, France) were of HPLC grade and used without further purification, and formic acid was from JT Backer (Ville, France). Tissue culture medium TC 199 (10 x concentrated with Earle's salts) and glutamine were purchased from Sigma Aldrich Chimie ( St Quentin Fallavier, France).
Gut sac preparation and incubation
Male Sprague Dawley rats (221-240 g weight, Depré, Saint Doulchard, France) were used in our experiments. Gut sacs were prepared and incubated with the target drugs at the stated concentrations as previously described (34-36). At the appropriate time points, sacs were removed, washed 3 times in saline and blotted dry, and the serosal contents collected. The sacs were digested individually in 25 ml of 1 M NaOH at 37°C for one hour, or in certain cases, homogenised in ultrapure water using an Ultra-turrax blender. The protein content of the digest or homogenate was measured by the method described by Peterson (37) with bovine serum albumin as standard. Samples of the mucosal and serosal fluids and the tissue digest were extracted for LC-MS analysis. From the volume of the sac contents and the quantity of each compound present, the appearance/transport was expressed as nanomoles or picomoles per mg of tissue protein.
Sample preparation
Midazolam (8.2 mg) was dissolved in 0.5 ml of methanol in a 125 ml flask and the volume was then completed with TC199 to give a 100µM solution. The final amount of methanol was 0.7 % (v/v) and 0.16 % (v/v) respectively for testosterone and midazolam in the solutions used for incubation.
The preparation of substrate solutions for incubation and the analytical procedures for testosterone, dextromethorphan and bupropion have been detailed in our earlier reports (34-36) for each compound. The analytical methods were all validated to quantify the substrates and their related metabolites in intestinal tissue after digestion in 1M NaOH. In 1 M NaOH, the extraction recovery was over 80% for midazolam and testosterone but it was only about 55% for dextromethorphan. As bupropion was not stable in 1M NaOH, the amount in the tissue was evaluated from a tissue homogenate. One ml of the homogenate was alkalinized with 20 µl of 1M NaOH and then extracted with 1 ml of diethylether. After manual agitation and centrifugation (5 min, 4000 x g), the organic layer was back extracted with 950 µl of water and 50 µl of formic acid before analysis of the aqueous layer (10µl). Extraction recoveries from homogenates were similar to those obtained in TC 199 (> 75%).
For testosterone, midazolam and dextromethorphan, the extraction procedure from intestine tissues was carried out after complete digestion of the tissue in 1M NaOH. In the experiments with testosterone, 0.5 ml of the resulting solution was neutralized with 43 µl of concentrated HCl, and 5 µl of progesterone (Internal Standard, 16 µM) and 0.375 ml of water were then added before extraction as previously described for serosal and mucosal media. After evaporation of the organic layer, the residue was dissolved in 1 ml of water and analysed by LC-MS, with the final concentration of Internal Standard (IS) at 0.8 µM.
For experiments with dextromethorphan, after complete digestion of the tissue in 1 M NaOH, 50 µl of Internal Standard (codeine, 10 µM) were added to 0.5 ml of the digest, then 28 µl of 1% acetic acid (pH 6) and 0.422 ml of water and the mixture extracted as previously reported for the serosal content (35). The residue was dissolved in 1 ml of water after evaporation of the organic layer and the final codeine concentration was 0.5µM.
For the midazolam experiments, 0.375 ml of TC 199 and 25 µl of bromazepam (6 µM) used as IS were added to 0.1 ml of serosal fluid, and the mixture was then extracted with 0.5 ml of a mixture of hexane/dichloromethane (1/1). After agitation, the organic layer was removed and evaporated to dryness under nitrogen and the residue dissolved in 0.1 ml of water for the quantification of midazolam and hydroxymidazolam. For the mucosal fluid, 50 µl was removed, diluted with 0.2 ml of TC 199 and added to 0.25 ml of the (IS) 6 µM bromazepam). Samples were then extracted as described for serosal fluid and after evaporation the residue was dissolved in 1 ml of water. The final concentration of the IS was 1.5 µM in all samples. When intestinal tissues were completely digested in 1M NaOH, 0.5 ml of the resulting solution was added to 25 µl of IS (6 µM) and 0.375 ml of water and acidified with 0.1 ml of acetic acid before extraction as previously described for serosal and mucosal media. After evaporation, the residue was dissolved in 1 ml of water before LC-MS analysis. The IS concentration was 1.5µM.
LC-MS Analysis
The LC-MS system consisted of a Waters 2690 separation module interfaced to a ZQ mass spectrometer equipped with an electrospray ionisation source (Waters, St Quentin, France). The apparatus was managed with a Masslynx software (Micromass, version 3.5). LC-MS analysis for testosterone, dextromethorphan and bupropion metabolism studies were run in positive mode as previously reported (34-36).
In experiments with midazolam, analyses were run in positive mode (electrospray) with capillary and cone voltages set to 3.5 KV and 35 V, the temperature of the heated capillary at 280° C and the nitrogen nebulizing gas flow at 360 L/h. The mobile phase consisted of a mixture of water/methanol/formic acid 1% (38/42/20) with a flow rate of 0.2 ml/min in isochratic conditions for a run time of 15 min. Quantifications were run in SIR mode by selecting the characteristic (M+H)+ ions: m/z = 326 for midazolam, m/z = 342 for 1-hydroxymidazolam and m/z = 315 for bromazepam used as internal standard at a concentration of 1.5 µM for all the quantifications. The retention times were 3.8 min, 5.8 min and 12.8 min respectively for midazolam, 1-hydroxymidazolam and bromazepam. Any matrix effect was detected by the analysis of TC199 without analytes after extraction.The linearity of the method was verified in the range of 0.025 µM to 3 µM for midazolam and hydroxymidazolam. Intra-day repeatability was determined by analysing, on the same day, a set of midazolam and 1-hydroxymidazolam solutions in TC199 medium at 0; 0.015; 0.025; 0.05; 0.25; 0.4; 1; 2; 3 µM. The solutions were analysed after extraction by the procedure previously described for sample preparation. Quality control (QC) solutions at three concentrations (0.06; 0.75 and 2.5 µM) were analysed three times each. The procedure used for intra-day repeatability was reproduced each day for five days to assess inter-day repeatability (n =12 assays). Intra-day standard deviation (RSD %) for the compounds assayed were between 3.5 % and 17.6 % for midazolam and between 3.1 % and 10.6 % for 1-hydroxymidazolam. Inter-day repeatability (RSD %) was between 5.3 % and 15.9 % for midazolam and between 4.5 % and 15.5 % for 1-hydroxymidazolam. Accuracy (% bias) was 10.56 % (0.06 µM); 4.89 % (0.75 µM) and 1.67 % (2.5µM) for midazolam and 2.50 % (0.06 µM), -2.44 % (0.75 µM) and -2.60 % (2.5 µM) for 1-hydroxymidazolam. Inter day accuracy (% bias ) was 7.64 % (0.06 µM); 7.56 % (0.75 µM) and 4.2 % (2.5 µM) for midazolam and 12.5 % (0.06 µM), -0.56% (0.75 µM) and -0.4 % (2.5 µM) for 1-hydroxymidazolam. The limits of quantification (LOQ, S/N > 10) were 0.0025 µM (4 ng injected) for midazolam and 0.025µM (40 ng injected) for 1-hydroxymidazolam respectively.
Stability
Calibration curves were constructed from stock solutions of substrates and metabolites stored at -20°C. Samples containing the different analytes prepared from the stock solutions were analysed periodically to verify the stability of the compounds. Stock solutions remained stable for all compounds for at least 4 months. Tissues were analysed immediately after complete digestion in 1 M NaOH, however the stability of testosterone, dextromethorphan and midazolam was satisfactory for 24 hours under these conditions.
RESULTS
Figure 1 shows the appearance of midazolam in the serosal compartment of the gut sac as a function of time after incubation of 100 µM midazolam in the mucosal media for 60 min. From the quantity of midazolam recovered in the serosal content of the gut sac, we have calculated the Apparent Permeability (Papp) as described previously (29) from the rate of uptake for 30 minutes. The value is shown in Table 1 along with the Papp values calculated for testosterone, dextromethorphan and bupropion in our previous studies in order to see the correlation between lipophilicity and absorption across the mucosa. Substrates were accumulated in the tissues, particularly midazolam and dextromethorphan which were the most lipophilic and were found to be 3 to 4 fold higher than the other compounds (Table 2).
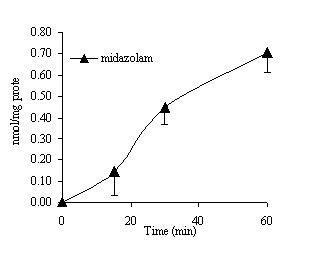
Figure 1. Appearance of midazolam in the serosal media during gut sac incubation with 100 µM midazolam (mean of 6 sacs ± SD).
Table 1. Apparent permeability (Papp) and lipophilicity (Log P) of compounds.
|
|
|
Midazolam |
2.08 ± 0.50 x 10-5 |
4.331 |
Bupropion |
5.68 ± 0.46x10-5 |
2.492 |
1calculated, 2experimental
Table 2. Amount of compounds accumulated in the gut tissues after 30 minutes of incubation.
|
Amount in tissue (n = 6) |
||
|
nmoles / mg protein |
% of the initial dose1 |
% of the initial dose |
Midazolam |
4.24 ± 0.08 |
0.43 ± 0.08 |
11.7 ± 2.4 |
Bupropion |
1.56 ± 0.18 |
0.14 ± 0.05 |
3.1 ± 0.7 |
1amount obtained in tissues calculated as a percentage of the dose incubated in the mucosal medium, expressed per mg protein.
As previously shown for the metabolites of testosterone, dextromethorphan and bupropion, 1-hydroxymidazolam, the related metabolite of midazolam was also detected in the serosal media as shown in Figure 2.
The amount of metabolites resulting from a specific CYP enzyme activity, found in the serosal content, was between 0.5-10 pmoles/mg protein, after 60 min. The quantity of 1-hydroxymidazolam (5.4 ± 1.2 pmoles/mg protein), 6-b-hydroxytestosterone (6.9 ± 3.1 pmoles/mg protein) and methoxymorphinan (2.2 ± 0.6 pmoles/mg protein), usually used as marker of CYP3A4/1 activity, were in the same order of magnitude in the serosal medium. Approximately twice as much hydroxybupropion (9.5 ± 1.2 pmoles/mg protein), which is characterized as a specific probe of CYP2B6 activity, was observed, whereas dextrorphan resulting from O-demethylation of dextromethorphan (CYP 2D6 activity) was obtained to a much lower extent (0.5 ± 0.1 pmoles/mg protein), in the serosal content after 60 min of incubation with the related substrate. After being formed in enterocytes, metabolites could accumulate in the serosal content or pass through the apical membrane towards the mucosal side either by passive diffusion or by the action of efflux transporters. Figure 3 shows the concentrations normalised to the sac size (mg protein) of the different metabolites on each side of the epithelial mucosa after incubation of the related substrates (100 µM) for 30 minutes.
It can be seen that the CYP3A4 metabolite of dextromethorphan, methoxymorphinan, was found at a higher concentration in the mucosal compartment than the other metabolites and than in its respective serosal compartment. This strongly suggested that it was a substrate for an efflux transporter. However, as shown in Figure 4, the co-incubation of the P-gp inhibitor verapamil (100 µM) with dextromethorphan (100 µM) in the mucosal media showed that verapamil had no effect on the transport of dextromethorphan or the metabolite methoxymorphinan. This would indicate that methoxymorphinan may be a substrate for efflux transporters other than P-gp.
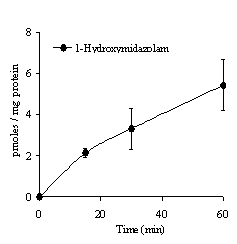
Figure 2. Appearance of 1-hydroxymidazolam in the serosal medium during gut sac incubation with 100 µM midazolam in the mucosal media (mean of 6 sacs ± SD).
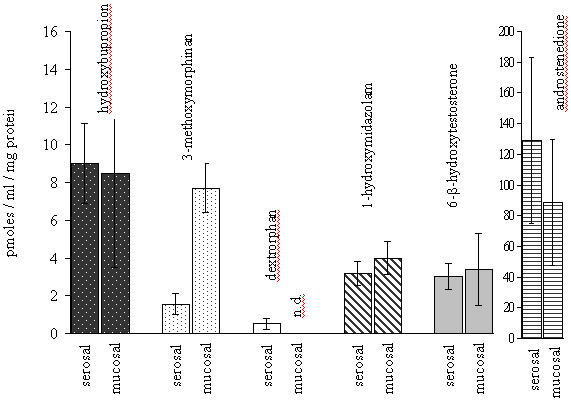
Figure 3.Normalised concentrations (pmoles/ml/mg protein) of metabolites on each side of the everted gut sac after 30 min of incubation (100 µM substrate). Each concentration is the mean of six measures : three sacs from two independent experiments, n.d.: not detected.
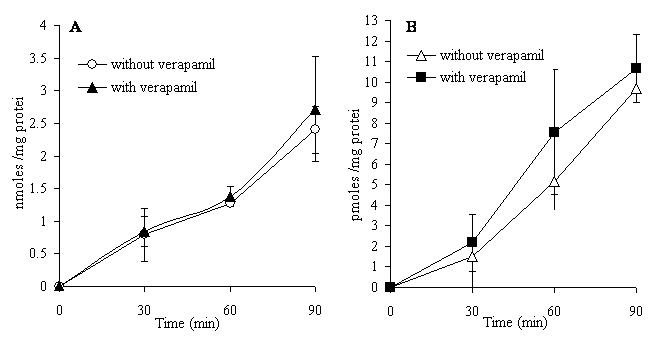
Figure 4. Accumulation of dextromethorphan (A) or 3-methoxymorphinan (B) in the serosal medium with or without verapamil (100 µM) co-incubated with 100 µM dextromethorphan in the mucosal media. Each point is the mean ± SD of four sacs of two independent experiments.
Discussion
The question of the intestinal first-pass metabolism has been the focus of increasing attention since the intestine was recognized as contributing substantially to drug metabolism due to the presence of the P450 family of oxidative enzymes. Seven P450 isoforms (CYP1A1, CYP2B1, CYP2C6, CYP2C11, CYP2D, CYP3A1 and CYP3A62) have been reported to be expressed in rat intestinal epithelial cells (38-42).
In spite of the various approaches used to study intestinal metabolism, there is still a need for simple in vitro models to study intestinal metabolism during the absorption of drugs by the intestine. The preparation of intestinal microsomes appears to be more difficult than for hepatic microsomes and the enzymatic activity of such microsomes depends considerably on the preparation technique and can be affected by the freezing (17,43). It is also difficult to extrapolate from the rates of metabolism obtained with isolated microsomes to the situation in vivo because the rate of metabolism is dependent on many physiological factors, not least, the rate of transport of drugs into the cells and the intactness of the whole cells. Dextromethorphan O-demethylation (dextrorphan formation) and hydroxybupropion formation are often chosen to monitor respectively the activity of CYP2D6 and 2B6 in man. Dextromethorphan can also be N-demethylated to give 3-methoxymorphinan as a result of the activity of CYP3A4, and thus is a probe for two different enzyme activities depending on the metabolites formed, whereas testosterone and midazolam are often used as markers of CYP3A4/1 activity. The formation of 1-hydroxymidazolam, after incubation of the sacs with 100µM midazolam, confirms the CYP3A4/1 enzyme activity in gut during the passage of midazolam across the epithelium. CYP3A activity was previously demonstrated for methadone (33) and testosterone (34) with the same model. In addition, dextromethorphan N-demethylation (3-methoxymorphinan production) also reflects CYP3A activity.
The rapid removal of the intestine from the animal and its immediate placing in TC 199 medium ensures its viability and maximum biochemical function. Caco-2 or other cell cultures have also been used to study the possible synergy between P-gp and CYP3A. However, Caco-2 cells lines do not express CYP3A4 which needs to be induced or transfected, thus giving a model with artificially created enzyme levels. The metabolism data given by the everted gut sac model are more comparable with the physiological situation as they are obtained from the interaction of substrates with the CYP enzymes inside enterocytes and during the passage through the cells. The catalytic activities of CYP450 enzymes in rat gut sacs were previously studied by Takemoto (43) who reported an activity of 83 ± 19 pmol/min/mg protein for testosterone 6-b-hydroxylation, which is high compared with our results. However, the system used was very different. The major difference is that we used TC 199 medium for the intestinal incubation while Takemoto used a simple phosphate buffer supplemented with a NADPH-generating system. In addition, Takemoto used an open system with an untied intestinal segment. Under these conditions, testosterone could access the enterocytes on each side of epithelium and it could partly explain why the amount of metabolite was higher. In addition, it is not possible to distinguish the amount of metabolites on the respective mucosal and serosal sides of the epithelium with an open segment .
To be able to separately assay the different compartments, mucosal, serosal and tissue is very important for the study of drug efflux and absorption in the intestine, particularly to evaluate the interplay between drug transport and metabolism. For example, P-gp and CYP3A4 have been postulated to work together to protect the body from the absorption of harmful xenobiotics including drugs (44). While synergism between P-gp and CYP3A4 has been suggested in recent years (25), direct evidence to support it remains elusive. In a attempt to answer the question of this interplay, a Caco-2 cell model where CYP3A was induced by vitamin D3, was used by Hockman et al (27). They noted that the main problem was to identify an appropriate in vitro model expressing both CYP3A and P-gp. As we have shown with the rat gut sac model, several CYP isoforms are active in enterocytes and metabolism and transport studies through the intestinal epithelium could be studied simultaneously. We have also demonstrated the activity of P-gp in gut sacs using digoxin as the substrate (32,45). Thus the everted sac may be a very useful tool to study the interplay between P-gp and metabolic enzymes.
Androstenedione was an oxidative product of testosterone which was itself rapidly hydroxylated (46). Two possible mechanisms have been proposed for androstenedione formation: one by spontaneous dehydration of the gem-diol 17a,17b-dihydroxy-androst-4-ene-3-one (47) and the other by direct oxidative dehydrogenation (46). This metabolite has not been systematically researched in testosterone metabolism studies, probably because of the lack of enzyme specificity in its formation and the rapid hydroxylation it undergoes. In our system, after incubation with testosterone, androstenedione was the major metabolite detected on both the serosal (172 ± 51 pmoles/mg protein at 60 min) and mucosal side of the epithelium, showing the strong metabolic activity of the gut. 6-b-hydroxytestosterone was quantified on both sides and the CYP2B1 activity was also demonstrated by the presence of 16-a and 16-b-hydroxytestosterone detected in the serosal and mucosal media. Recent studies of midazolam and testosterone metabolism with rat intestinal tissue slices also reported the formation of 1-hydroxymidazolam and 6-b, 16-b, 16-a hyroxytestosterone and androstenedione respectively (48).
After being formed in the enterocytes, drug metabolites could cross the basolateral membranes of the cells into the serosal compartment or cross the apical membrane to the mucosal side. While the presence of metabolites in the mucosal medium could be due to simple transmembrane diffusion, it may also be indicative of the activity of the well-documented efflux transport mechanisms in the apical membrane. P-glycoprotein is the most studied of the efflux transporters and has a very broad substrate specificity, although other transporters such as MRP2 (multidrug resistance protein 2) and BCRP (breast cancer resistance protein) may also be important in xenobiotic efflux from the enterocytes. After studying the effect of grapefruit juice on dextromethorphan pharmacokinetics in man, Di Marco et al. (49) hypothesized that dextromethorphan is a substrate of P-glycoprotein (P-gp) or a related membrane efflux protein of the gastrointestinal tract. We have not found any reports in the literature to corroborate this hypothesis. When we studied dextromethorphan absorption and metabolism with the everted gut sac, we observed an accumulation of 3-methoxymorphinan in the mucosal medium with a three fold higher concentration than in the serosal side. This may be the result of the efflux from the mucosal cells by an active process such as the transporter P-gp. However, the transport of dextromethorphan and this metabolite were not modified in the presence of verapamil, a P-gp inhibitor, thus demonstrating that dextromethorphan and methoxymorphinan were not P-gp substrates and suggesting that other efflux transporters may be implicated. The presence of metabolites in both the serosal and mucosal compartments was also detected with other substrates and could be explained by a simple diffusion of metabolites across the cell membranes until reaching a balance on both sides.
These results do not agreed with those of Engman et al.(18) who found that the concentration of 6-b-hydroxytestosterone was two fold higher on the apical side than the basolateral side of Caco-2 cells treated with 1a-25-dihydroxy vitamin D3. However, it is difficult to compare these two systems because in the Caco-2 cells, CYP3A-mediated metabolism was induced by the vitamin D3 and testosterone was applied on the both sides of the cell monolayer.
The everted gut sac method is a useful tool to quantify and identify the metabolism of candidate drugs during their absorption and we have shown that the CYP isoforms of 3A, 2B and 2D families are active in the mucosal cells of the rat intestine. Given that the rat is a widely used species for the pre-clinical assessment of drug absorption and metabolism, such studies are very valuable in evaluating the role of mucosal drug metabolism by different enzymes in oral drug bioavailability. In addition this model offers the possibility to quantify the proportion of compounds accumulated in the intestinal cells which is not taken into account in most cell culture models. The model also has considerable potential for studying the interplay between transporters and metabolism by using specific substrates and inhibitors and such studies are now in progress in our laboratory.