J Pharm Pharmaceut Sci (www.cspscanada.org) 10(1):86-128, 2007
Perspectives of Biodegradable Natural Polysaccharides for Site-Specific Drug Delivery to the Colon
Anekant Jain, Yashwant Gupta and Sanjay K. Jain
Pharmaceutics Research Projects Laboratory, Department of Pharmaceutical Sciences, Dr. Hari Singh Gour University, Sagar-470 003 (MP). INDIA.
Received January 15, 2007; Revised, April 4, 2007; Accepted, April 15, 2007; Published, April 18, 2007.
Corresponding Author:Sanjay K. Jain, Pharmaceutics Research Projects Laboratory, Department of Pharmaceutical Sciences, Dr. Hari Singh Gour University, Sagar-470 003 (MP). INDIA. E-mail: drskjainsagar@rediffmail.com
ABSTRACT - The ability to deliver drugs to the human colon in a specific manner has become feasible over the years. Targeting pharmaceutical drugs to the colon makes it possible to achieve local or systemic drug delivery to this site. To deliver the compounds in a non-degraded form to the lower part of the gastrointestinal tract, they must first of all pass through the stomach, the upper part of the intestine and must use the characteristics of the colon to specifically release the drugs in this part of the digestive tract. Among the different approaches available to achieve targeted drug release to the colon, the use of especially biodegradable polymers holds great promise. This family of natural polymers has great appeal to drug delivery as it is comprised of polymers with a large number of derivatizable groups, a wide range of molecular weights, varying chemical compositions, and, for the most part, low toxicity and biodegradability yet high stability. The most favorable property of these materials is their approval as pharmaceutical excipients. Polysaccharidases are bacterial enzymes that are available in sufficient quantity to be exploited in colon targeting. Bacterial sources of polysaccharidases as well as a detailed treatise of the enzymatic flora of the colonic region is reviewed, followed by a wide range of the polysaccharides which can be used solely for the purpose of colon-specific drug delivery. Present article also discusses few of the mechanisms by which members of the intestinal microbiota degrade complex polysaccharides. A final overview of the various approaches to target drugs to the colon utilizing natural polysaccharides, the limits and the future developments in this field with these natural polymers is discussed which leads to the conclusion that polysaccharides show high potential in achieving colon-specific drug delivery
1. Introduction
The advantages and necessity of colon targeting to provide more effective therapy for colon related diseases, such as irritable bowel syndrome, colon cancer [1] and inflammatory bowel disease (IBD), including Crohn’s disease and ulcerative colitis [2], have been well recognized. The colon, as a site for drug delivery, offers distinct advantages on account of a near neutral pH, a much longer transit time, relatively low proteolytic enzyme activity, and a much greater responsiveness to absorption enhancers [3]. These criteria favor this distal part of the gastrointestinal tract (GIT) as a site for the delivery of various drug molecules, including proteins and peptides [4]. Colon-specific delivery systems should prevent the release of the drug in the upper-part of GIT and require a triggering mechanism to affect an abrupt release on reaching the colon. In the past, various primary approaches for colon-specific delivery, such as pro-drugs, pH sensitive polymers, timed release delivery systems, and microbially degraded delivery systems, have achieved limited success. The majority of these systems, developed during the past decade, were based on pH and time dependent mechanisms with limited in-vivo evaluation. Minor variation in pH between the small intestine and the colon makes the pH-dependent systems less specific, in terms of targeted release in the colon. Time-dependent formulations predominantly depend on the transit time of the delivery system in the GIT. A major limitation with these systems is that in vivo variation of the small intestinal transit time may lead to release of the bioactive in the small intestine or terminal part of the colon. The patho physiological state of an individual will have a significant impact on the performance of these time-dependent systems. Patients with irritable bowel syndrome [5] and ulcerative colitis [6] exhibited accelerated transit through different regions of the colon.
However, this review will focus on the application of polysaccharides for colon-specific drug delivery with emphasis on polysaccharide degradation by the enzymes present in the colon and their nature and regulation of nutrient exchange between mammalian hosts and intestinal microorganism affecting biodegradation. Attempt has also been made to focus on recent insights about the molecular mechanisms of few of the polysaccharides by which intestinal bacteria help the host degrade dietary carbohydrates and how they impact the host’s ability to absorb and metabolize these and other nutrients.
1.2 COLONIC DRUG DELIVERY
Site-specific drug delivery to the colon has attracted considerable attention for the past few years in order to develop drug delivery systems that are able to release drugs specifically in the colon in a predictable and reproducible manner. Colonic drug delivery in general has been reviewed by Basit [7] and Chourasia and Jain [8]. A chronic inflammatory disease such as ulcerative colitis requires daily treatment with aminosalicylates, and in some cases even daily treatments with corticosteroids such as prednisolone [9]. The site specific drug delivery to colon is important for the treatment of diseases associated with the colon, reducing the side effects of the drug and reducing the administered dose. The pharmaceutical formulations available for this treatment are slow release oral formulations or enemas and foams for rectal administration. The formulations, however, are not truly site-specific, and the treatments are connected with a high frequency of systemic side effects. The side effects appear to be more frequent after administration of oral formulations compared with rectal formulations because of the large degree of systemic absorption from the upper gastrointestinal tract. Drugs for which the colon might appear as a potential absorption site (e.g., peptides and vaccines) may also advantageously be delivered to this region for systemic absorption because, the enzymes which are responsible for the degradation of the peptides or vaccines are significantly present in the upper part of the GIT. Relevant drugs for colon-specific associated diseases are listed in Table 1 [10] and presented diagrammatically in Figure 1 [11].
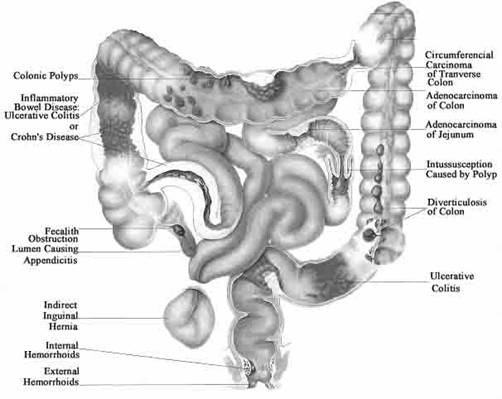
Figure 1: Diagrammatical presentation of Colon associated diseases11.
Table 1. Colon targeting diseases, drugs and sites10
Target Sites |
Disease Conditions |
Symptoms |
Drugs and active agents |
Topical / Local action |
Inflammatory Bowel Disease |
||
Crohn’s disease |
Diarrhea, Abdominal pain and cramping, blood in stool, ulcers, reduced appetite and weight loss |
Hydrocortisone, Budenoside, Prednisolone, Sulfasalazine, Olsalazine, Mesalazine and Balsalazide, Infliximab
|
|
Ulcerative colitis |
Inflammation in the rectum, rectal bleeding, rectal pain
|
5-Amino salicylic acid, Sulfasalazine, Balsalazide, Infliximab, Azathioprine and mercaptopurine
|
|
Irritable bowel syndrome |
Abdominal pain or cramping, a bloated feeling, flatulence, diarrhea or constipation — people with IBS may also experience alternating bouts of constipation and diarrhea, mucus in the stool
|
Dicyclomine, Hyoscine, Propantheline, Cimetropium, Mebeverine, Trimebutine, Scopolamine, Alosetron, Tegaserod |
|
Colorectal cancer |
A change in bowel habits, narrow stools, rectal bleeding or blood in stool, persistent abdominal discomfort, such as cramps, gas or pain, abdominal pain with a bowel movement, unexplained weight loss |
5-Flourouracil, Leucovorin, Oxaliplatin, Irinotecan, bevacizumab and cetuximab |
|
Diverticulitis
|
Formation of pouches (diverticula) on the outside of the colon due to bacterial infection
|
Bactrim |
|
Antibiotic associated colitis |
Overgrowth of Clostridium difficile and its subsequent toxin production |
clindamycin, broad-spectrum penicillins (eg, ampicillin, amoxicillin), and cephalosporins |
|
| Hirschsprung's disease | Severe form of constipation in which bowel movement occurs only once or twice a week | Metronidazole, Vancomycin, Loperamide, Botulinum toxin | |
| Systemic action | To prevent first pass metabolism of orally ingested drugs |
---- |
Steroids |
Oral delivery of peptides |
---- |
Insulin |
|
Oral delivery of vaccines |
---- |
Typhoid |
|
Ulcerative colitis |
Ulcerative proctitis, pancolitis, fulminant colitis
|
Prednisolone metasulfobenzoate, tixocortol pivalate, fluticasone propionate, beclomethasone dipropionate and budesonide, Azathiorpine/6-mercaptopurine, Cyclosporin A |
|
Colon-specific drug delivery has been approached by a number of methods exploiting changes in the physiological parameters along the gastrointestinal tract [12-14]. The GIT transit time was utilized to formulate colon specific drug delivery systems which are so designed that their drug release is delayed to the time required for transiting drug from mouth cavity to distal part of small intestine i.e., ileum and subsequently drug release in the colon [13,15-17]. Factors influencing the transit time of pharmaceutical dosage forms in the various regions of the gastrointestinal tract appear to depend on diet, gastrointestinal motility, physical activity of the person, fasted or fed state of the person [18,19]. The change in pH along the gastrointestinal tract was also used to develop colon specific drug delivery systems by applying coatings that were intact at low pH and dissolved at neutral pH. The pH of the colon is, however, often lower than the pH of the small intestine, which in turn may be as high as 8 or 9, resulting in a too early release of a drug [17]. Other reports conclude that pH is useful as a trigger for drug release in the colon [20,21].
A number of specific regional characters of the colon can be explored for site specific drug delivery to the colon. In the colon, an extensive growth of anaerobic microorganisms is observed [14,10]. In contrast, the microflora of the upper gastrointestinal tract is not as prominent, and consists mainly of aerobic microorganisms. These colonic microflora produce a large number of hydrolytic [22] as well as reductive enzymes [23] which can potentially be utilized for colon-specific drug delivery. Prodrugs [24] and coatings based on azoaromatic polymer [25] and matrices [26] containing azoaromatic cross-links are examples of systems that potentially are degradable by reductive enzymes released by colonic bacteria [10]. Apart from azo reductase enzyme release other polysaccharidases like glucosidases; glycosidases are also released by colonic microflora, which are responsible for the degradation of polysaccharides [27,28]. Hence, drug delivery systems based on polysaccharide can also be used for colon specific drug delivery.
1.2 SIGNIFICANCE OF COLONIC DISEASES FOR DRUG TARGETING
The mucus and epithelium membranes represent crucial physical barriers to the uptake of peptides and proteins from the normal healthy intestinal lumen in to the mucosa. Variations in these layers during disease may have important implications for drug delivery to affected regions. In addition to alterations in the colonic mucus layer being affected by number of diseases, the morphology of the mucosa may also be influenced.
The permeability of the intestinal mucosa is increased in most patients with Crohn’s disease and 10-20% of their clinically healthy relatives. Permeability is also increased in individuals with celiac disease, trauma, burns and administered non steroidal anti-inflammatory drugs. This increased permeability is attributed to loosening of tight junction [29]. In addition, inflammatory diseases of the bowel, including ulcerative colitis [30] and Crohn’s disease [31], are also associated with epithelium inflammation and damage. A break in the continuity of the epithelial surface (whether as a result of tight junction relaxation or a break in the epithelium layer) may offer the opportunity for direct uptake in to the affected tissue of not only chemotherapies, but also pathological agents.
Abnormal epithelium cell development may lead to benign or malignant growths extending from the colonic epithelium [32]. It is interesting to speculate that the protruding nature of these growths (including polyps) may increase their exposure to the lumen contents and offer a unique opportunity for confrontation with orally administered drugs.
Age has significant impact on the health of the colon and consequently the requirement for therapy. The incidence of colorectal cancer increases with age [32]. Motility disorders of the GI tract (such as IBD) also increase with age. It has been proposed that age-associated changes in the levels of neuroendocrine peptides (regulators of GI-secretion, absorption, motility, cell proliferation, local immune defense, and blood flow) may affect the health status of the colon [33]. From the perspective of therapy, the preferential distribution of non-infective diseases of the colon is particularly important. With the exception of benign colonic carcinoma and Crohn’s disease (a disease not restricted to large intestine [34], noninfective diseases predominate in the left side of the descending colon and in the sigmoid/rectum. Diseases that dominate this region include large bowel cancer [31], diverticular disease [35], irritable bowel syndrome, IBS [36] and ulcerative colitis [34]. This preferential distribution is an important consideration for drug targeting strategies. Access to the descending colon would be expected to be more direct through the rectum than through the mouth. However, the ease of oral drug consumption offers a more attractive form of administration than any other alternatives.
1.3 COLONIC DRAINAGE
The rate of blood flow to and from the absorptive epithelium is likely to determine the rate of drug absorption from the lumen. Blood flow to the colon is less than to small intestine, and the proximal colon receives a more prolific supply than the more distal regions [37]. The nature of the drainage from the colon determines the fate of a drug taken up from this region. The drainage from the upper colon appears distinct from that of the more distal regions of the large intestine, where the former is drained by both the hepatic portal veins and the lymphatics and the latter is drained predominantly by the lymphatics [38].
The blood flow rate in humans is 1000-2000 times greater than the rate of lymphatic flow [39], and blood circulation has consequently been considered more significant for drug distribution [40]. However, drugs entering the blood through the hepatic portal vein circulate directly to the liver, where they may undergo rapid biotransformation (hepatic first-pass metabolism) before they are able to act. Introduction of drugs in to the lymphatic, on the other hand, has been considered a means of improving oral bioavailability, because drugs could potentially passage from the lymphatic in to the blood circulation before reaching to the liver [41]. The mode of uptake into the lymphatics may be through the M-cells or the epithelial cells [42].
1.4 INTESTINAL METABOLISM OF POLYSACCHARIDES
The microbial ecology of the intestine exhibits an astonishing degree of spatial and temporal complexity that defining microflora as “normal” is a daunting task. At least 500 bacterial species colonize the adult intestine, with 30–40 species comprising up to 99% of the total population [43-45]. If the average genome size and gene density of the species represented in the gut microbiota are similar to those in Escherichia coli (5 mega base pairs, 4000 genes), the microbiome would have a complexity of 2.5 giga base pairs and contain 2 million genes. The true extent of biodiversity in the gut is not known because most organisms cannot be cultured ex vivo. The intestinal tract is sterile at birth. The composition of the human gut microflora undergoes dramatic changes during postnatal development [46]. Despite the variations in the composition of the microbiota between and within individuals, there is a “functional stability” that allows the microbiota to maintain its capacity to carry out a basic set of biochemical reactions, including degradation of carbohydrates, synthesis of vitamins and fermentation.
1.4.1 Microflora of the Human Gastrointestinal Tract
Among the different approaches previously discussed, microflora activated systems appear to be more promising, since the abrupt increase of the bacterial population and associated enzymatic activity in the colon represents a non-continuous event, independent of the GI transit time [47,48]. Microbially activated delivery systems for colon targeting are being developed to exploit the potential of the specific nature of diverse and luxuriant microflora associated with the colon compared to other parts of the GIT. This vast microflora in the colon survives by fermenting the various types of substrates that have been left undigested in the small intestine [49]. The microflora of the human gastrointestinal tract consists of a coexisting mixture of aerobic, facultative anaerobic and strict anaerobic bacteria in a complex ecosystem. Table 2 shows the presence of microorganisms along the GIT of different animals. These bacteria produce a wide range of enzymes, such as β-glucuronidase, β-xylosidase, α-arabinosidase, β-galactosidase, nitroreductase, azoreductase, deaminase, urea hydroxylase etc [50].
The stomach and duodenum contain only few bacteria, all aerobic. Going down the gastrointestinal tract the total bacterial count increases, and in colon and faeces, the number of anaerobic bacteria is very high. Factors such as the short transit time of material through the stomach and small intestine and the low pH of the stomach are most likely responsible for the low bacterial content of these organs. In contrast, the long transit time of the colon, together with the high content of nutrients, allows colonization of about 400 different bacterial species in the colon. The predominant species of anaerobic bacteria in the colon are the bacteroides, bifidobacteria, eubacteria, clostridia, and gram-positive cocci [14]. The facultative bacteria are responsible for the low redox potential in the colon by their consumption of oxygen [51]. Changes in redox potential induced by the vast microflora of the colon have been used as a highly specific mechanism for drug targeting. Azo group containing polymers and their reduction by azoreductases produced by the colonic microflora have been exploited for the colon-targeted delivery of drugs [52, 53].
Factors responsible to influence the intestinal microflora are discussed by Rowland [23]. These factors are, however, of varying importance. The composition of the human flora was, for example, shown to be practically independent of diet, age, and geographic location [54-56] even though observations on varying concentrations of bifidobacteria and eubacteria in persons living on different diets have been reported [56]. On account of these variations, it should not be a serious problem with interindividual enzyme activities in humans. On the other hand, the intraindividual variation arising from periodic consumption of certain foods and drugs has to be considered seriously if the microflora should be utilized. Diseases, or treatment with antibiotics, may influence the microbial flora [57,58]. A disease resulting in decreased secretion of hydrochloric acid in the stomach, hypochlorhydria, may result in increased bacterial growth in the stomach [59]. Some microorganisms were found to be suppressed after treatment with antibiotic [58]. Different animal species and humans do not have the same microflora composition in the gastrointestinal tract (Table 2).
Table 2. The Composition of the Intestinal Flora of Four Animal Species and Man13
Site & test |
|
Mean log,, viable countb of the stated organisms |
|||||||
|
Entero-bacteria |
Entero- |
Lacto- |
Veil- |
Yeasts |
Clostridia |
Bacter- |
Bifido- |
|
Stomach |
|||||||||
Rabbit |
(3) |
Nc |
N |
N |
N |
N |
N |
5.7 |
5.5 |
Guinea-pig |
(9) |
N |
N |
3.9 |
5.7 |
N |
3.3 |
N |
5.9 |
Rat |
(8) |
4.1 |
3.9 |
6.6 |
7.8 |
4.1 |
3.3 |
7.0 |
7.6 |
Mouse |
(3) |
3.1 |
2.9 |
8.3 |
N |
6.3 |
3.6 |
8.1 |
8.3 |
Man |
(30) |
N |
N |
N |
N |
N |
N |
N |
N |
Upper small intestine |
|||||||||
Rabbit |
(3) |
N |
N |
N |
N |
N |
N |
4.5 |
4.4 |
Guinea-pig |
(9) |
N |
N |
3.5 |
N |
N |
3.2 |
N |
5.1 |
Rat |
(14) |
4.5 |
4.7 |
6.7 |
4.5 |
4.0 |
3.0 |
6.3 |
6.7 |
Mouse |
(3) |
4.2 |
4.5 |
7.7 |
N |
6.8 |
2.3 |
6.6 |
7.0 |
Man |
(30) |
N |
N |
1.0 |
N |
1.0 |
N |
2.5 |
2.0 |
Lower small intestine |
|||||||||
Rabbit |
(3) |
N |
N |
N |
N |
N |
4.7 |
5.4 |
6.1 |
Guinea-pig |
(9) |
N |
N |
4.0 |
N |
N |
3.4 |
6.3 |
6.0 |
Rat |
(8) |
4.1 |
5.0 |
6.8 |
5.1 |
3.9 |
3.3 |
6.9 |
7.6 |
Mouse |
(3) |
3.6 |
4.8 |
7.5 |
5.3 |
5.4 |
3.3 |
7.7 |
7.9 |
Man |
(5) |
3.3 |
2.3 |
N |
N |
2.3 |
N |
3.5 |
4.0 |
Large Intestine |
|||||||||
Rabbit |
(3) |
3.4 |
N |
N |
N |
N |
3.8 |
8.0 |
4.5 |
Guinea-pig |
(9) |
N |
3.2 |
4.3 |
4.0 |
N |
3.1 |
7.1 |
8.4 |
Rat |
(8) |
5.5 |
6.0 |
7.6 |
5.7 |
5.9 |
3.1 |
8.0 |
8.2 |
Mouse |
(3) |
5.7 |
5.2 |
7.4 |
6.3 |
6.8 |
3.3 |
8.3 |
8.2 |
Man |
(11) |
7.0 |
7.0 |
6.5 |
3.0 |
N |
N |
8.0 |
7.0 |
Rectum/Feces |
|||||||||
Rabbit |
(3) |
2.3 |
2.3 |
4.0 |
N |
N |
3.8 |
7.8 |
7.7 |
Guinea-pig |
(9) |
2.9 |
3.1 |
3.9 |
4.0 |
4.0 |
3.5 |
7.8 |
8.4 |
Rat |
(8) |
5.9 |
6.6 |
7.8 |
5.8 |
5.8 |
3.0 |
8.2 |
8.6 |
Mouse |
(3) |
5.0 |
4.8 |
8.1 |
6.6 |
6.6 |
3.3 |
8.4 |
7.8 |
Man |
(30) |
6.0 |
3.5 |
4.0 |
3.0 |
1.0 |
3.0 |
10.5 |
10.5 |
|
|||||||||
Table 3. Glycosidase Activities on Mixed Populations of Human Faecal Bacteria After 24 h Growth on Different Polysaccharide Substrates91.
Glycosidase |
Glycosidase activitya (nmol pnitrophenol released h-1 mg protein) |
|||
Starch culture |
Arabinogalactan |
Pectin culture |
Xylan culture |
|
a-L-Arabinofuranosidase |
1207 |
1054 |
963 |
764 |
b-D-Xylosidase |
1168 |
1036 |
9093 |
788 |
a-D-Galactosidase |
910 |
200 |
900 |
620 |
b-D-Galactosidase |
1197 |
1095 |
947 |
739 |
b-D-Fuosidase |
128 |
128 |
71 |
129 |
b-D-Galacturonidase |
11 |
ND |
ND |
120 |
b-D-Glucuronidase |
70 |
51 |
ND |
ND |
a-D-Glucosidase |
1149 |
1020 |
947 |
681 |
b-D-Glucosidase |
1001 |
859 |
769 |
572 |
a-D-Mannosidase |
ND |
ND |
ND |
19 |
b-D-Mannosidase |
58 |
50 |
ND |
38 |
|
|
|
|
|
ND, not detected. a Results are the mean data obtained form separate experiments in which faecal samples from 2 different persons were used. |
||||
Due to the toxicity associated with the synthetic polymeric systems, a wide variety of natural polymers are being used for the design and development of colon-targeted delivery systems. Most of these systems are based on the knowledge that anaerobic bacteria in the colon are able to recognize the various substrates and degrade them with the enzymes. The application of biodegradable natural polymers, which are resistant to degradation in the upper GIT (above the colon), has gained tremendous importance in pre-biotic food systems. Most of the recent research includes natural polysaccharides, especially from plant origin, being applied to create degradable colon-specific substrates. A number of colon-targeted delivery systems based both on polysaccharides and synthetic polymers, such as acrylics, have been designed and developed by various research groups [60-62]. In the current survey on colon-targeted delivery systems, an attempt has been made to review most of the polysaccharide systems with minimal chemical modification reported till date on colon targeting. As these polymers are fermented by the colonic microflora, they are categorized under ‘generally regarded as safe’ (GRAS). These polymers may also be utilized for the development of delivery systems for food/nutraceutical applications. From this it can be concluded that it is important to select an in vitro or in vivo model that mimics the conditions in humans the most. In some mammals (e.g., rat and mouse) the stomach and small intestine can contain considerable amounts of bacteria, depending on diet [63].
1.4.2 MICROBIAL CONTRIBUTIONS TO CARBOHYDRATE METABOLISM
Carbohydrates form the bulk of most human and animal diets and thus are critical nutrients for both host and microbiota. Mammals are well equipped to absorb simple sugars such as glucose and galactose in the proximal portion of their small intestine [64]. They can hydrolyze certain disaccharides (e.g., sucrose, lactose, and maltose) to their constituent monosaccharides. They can also degrade starch to glucose monomers but are generally limited in their capacity to hydrolyze and utilize other polysaccharides. Therefore, a large quantity of undigested dietary carbohydrate passes into the distal portion of the gastrointestinal tract each day. This material includes polysaccharides (including cellulose, xylan, and pectin) as well as undigested starch. The host’s problem of gaining nutritional benefit from these polysaccharides has been solved, in part, by adopting a microbial population that has the ability to degrade these biomolecules. A very efficient, mutually beneficial arrangement has evolved from a nutritional perspective (Figure 2). In the proximal gut, bacterial competition for absorbable monosaccharides could be detrimental to the host, but in nonruminants colonization is sparse in this region. Microbial digestion of carbohydrate polymers that reach the distal gut could benefit the host by helping it extract nutrient value from otherwise poorly utilized dietary substrates. Correspondingly, the distal regions of the gut are heavily colonized [45].

Figure 2: Overview of host and bacterial contributions to carbohydrate utilization in the intestine. Mammals absorb simple sugars, such as glucose and galactose, via active transport in the proximal regions of their small intestine. However, they have limited intrinsic capacity to digest dietary polysaccharides. Undigested polysaccharides such as cellulose, xylan, and undigested starch, as well as host-derived glycans (mucins and glycosphingolipids) pass into the distal regions of the small intestine (ileum) and colon where they are degraded by resident microbes. Bacteria ferment the resulting monosaccharides, and the byproducts of this fermentation (short chain fatty acids) are absorbed and utilized by the host63.
1.4.3 MICROBIAL DEGRADATION OF STARCH
Members of the genus Bacteroides are among the most abundantly represented species in the distal human intestine [65]. This group of anaerobes can degrade and ferment a wide variety of polysaccharides, including xylan [66], psyllium hydrocolloid [67], and numerous other plant polysaccharides [68]. Such saccharolytic capacity may explain, at least in part, why Bacteroides species are such successful gut colonizers. An analysis of the molecular details of how bacteroides utilize these polysaccharides provides a useful paradigm for understanding how intestinal microbes sense and exploit nutrients in their environment. The starch utilization system (sus) of Bacteroides thetaiotaomicron is the best understood example of polysaccharide utilization. B. thetaiotaomicron is a prominent, genetically manipulatable member of the normal mouse and human distal intestinal microflora [65,69,70]. Eight B. thetaiotaomicron genes have been identified that participate in starch metabolism (Figure 3) [71-73]. Seven of these genes (susA–susG) encode proteins that mediate the initial utilization steps [74]. SusC, SusD, SusE, and SusF are all outer membrane proteins that interact to form a complex that mediates binding of starch to the bacterial cell surface [71,74]. Bound starch is then hydrolyzed by α-amylases encoded by susA and susG. The products of these genes cleave starch at α 1,4 and α 1,6 linkages to yield smaller oligosaccharides. SusG is an outer membrane protein, whereas SusA is soluble and is located in the periplasmic space. These two proteins act in concert with an α-glucosidase (SusB) that breaks down their oligosaccharide products to glucose monomers [72]. By binding starch to its outer membrane prior to initiating hydrolysis, B. thetaiotaomicron can degrade a large polymer into oligosaccharides small enough to pass through outer membrane porins without losing these digestion products to nearby microbial competitors [75]. An eighth gene, susR, encodes a transcriptional activator that responds to the presence of maltose or larger oligosaccharides by increasing transcription of susA–susG [73]. Thus, the starch binding and degrading activities in B. thetaiotaomicron are expressed only if cells are exposed to glucose oligomers. This arrangement is not unique. In this and other examples discussed below bacteria expend energy to express genes involved in nutrient metabolism only when the particular nutrient is available in the gut ecosystem.
1.4.4 MICROBIAL DEGRADATION OF HOST-DERIVED GLYCANS
In addition to their ability to hydrolyze numerous plant polysaccharides, Bacteroides species have also evolved the capacity to degrade a variety of host-derived glycoconjugates (glycans) including chondroitin sulfate [76,77], mucin [68], hyaluronate [78], and heparin [79]. The chondroitin sulfate and hyaluronate degradation pathways in B. thetaiotaomicron (Figure 4) have features resembling those of the starch utilization system [80]. As with starch, chrondrotin sulfate and hyaluronate are too large to enter the cell through porins. There is evidence that both of these host glycans are first bound to the bacterial cell surface prior to degradation. csuF encodes an outer membrane protein that is a candidate receptor for these glycoconjugates: a transposon insertion in csuF abrogates growth on intact chondroitin sulfate or hyaluronate but does not disrupt growth on their disaccharide components [80]. B. thetaiotaomicron degrades chondroitin sulfate and hyaluronic acid to disaccharides via the action of two chondroitin lyases, both of which are located in the periplasm. The b-glucuronidase that cleaves the resulting disaccharides to monosaccharides is located in the cytoplasm [81]. The chondroitin sulfate and heparin utilization pathways are biochemically distinct. However, they are linked by chuR, which controls expression of the genes involved in utilization of both glycans [79]. Like components of the starch utilization pathway, the hydrolases involved in breakdown of chondroitin sulfate and heparin are inducible: they are only produced when B. thetaiotaomicron is grown in the presence of chondroitin sulfate or heparin, or in the presence of their component disaccharides [76]. B. thetaiotaomicron’s ability to utilize these host glycans appears to be critical for its survival in the intestinal ecosystem. For example, an insertion in chuR, which abolishes growth on both chondroitin and heparin, prevents the mutant strain from successfully competing with an isogenic wild-type strain in cocolonization studies of germ-free intestines [78]. Mucins and glycosphingolipids represent two other classes of host glycans degraded by secreted bacterial hydrolases. Mucins are high molecular weight, heavily glycosylated glycoproteins produced by goblet cells. They serve the host by helping to maintain the integrity of the mucosal barrier [82]. Indigenous bacteria have evolved a number of enzymes for breaking down and utilizing the oligosaccharide side chains of host mucins [83]. These side chains typically have very heterogeneous and complex structures, with diverse monosaccharide components connected by a variety of glycosidic linkages. Mucin degradation frequently requires the participation of several bacterial species, each of which expresses some but not all of the required glycosidases. However, some members of the Bacteroides, Ruminococcus, and Bifidobacterium genera are able to support complete side chain degradation [84].

Figure 3: Model of Bacteroides thetaiotaomicron starch utilization. Eight starch utilization genes have been identified. SusC–SusF are outer membrane proteins that form a complex that mediates binding of starch to the bacterial cell surface. Bound starch is then digested by µ-amylases encoded by susA and susG. SusB is an glucosidase that breaks down oligosaccharides released by SusA and SusG into glucose. SusR encodes a transcriptional activator that binds to maltose or larger oligosaccharides and increases transcription of susA–susG71-73. ["Reprinted, with permission, from the Annual Review of Nutrition, Volume 22 (c)2002 by Annual Reviews: www.annualreviews.org"].
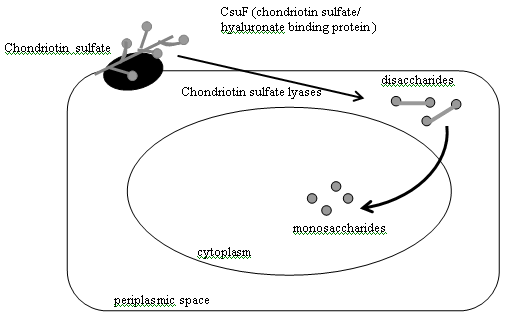
Figure 4: Model of Bacteroides thetaiotaomicron chondroitin sulfate/hyaluronate utilization. Both chondroitin sulfate and hyaluronate are likely first bound to the bacterial outer membrane prior to degradation. CsuF, an outer membrane protein, is a candidate receptor. Chondroitin sulfate and hyaluronic acid are degraded to disaccharides in the periplasm by chondroitin lyases I and II. A cytoplasmic b-glucuronidase cleaves the disaccharides to yield monosaccharides. These three hydrolases are induced only when B. thetaiotaomicron is grown in the presence of chondroitin sulfate or hyaluronate76,80. ["Reprinted, with permission, from the Annual Review of Nutrition, Volume 22 (c)2002 by Annual Reviews: www.annualreviews.org"].
Glycosphingolipids constitute another group of host glycans that can be degraded by secreted bacterial glycosidases [85,86]. These glycans are found on gut epithelial cells and have oligosaccharide side-chains attached to a lipid ceramide group. Glycosphingolipids on shed epithelial cells are progressively degraded in the lumen of the intestine [87]. There is evidence of an in vivo selection for bacteria that are able to hydrolyze the oligosaccharide chains of glycosphingolipids produced by the individuals colonization. Hoskins & Boulding found that the fecal flora of adult humans with the histo-blood group A phenotype degrade A but not B antigens, whereas the fecal flora of individuals with the histoblood group B phenotype degrade B but not A [88,89]. Members of the Bifidobacterium and Ruminococcus genera were responsible for this activity of their assay system. In summary, host glycans appear to be a critical nutrient source for B. thetaiotaomicron and other bacterial species. The advantages of being able to utilize these glycans are readily apparent: they are constantly replenished due to epithelial cell turnover; once degraded they are readily fermented to yield carbon and energy; and competition for these glycans is limited, because they are degraded only by some bacterial species [90].
1.5 Polysaccharide Fermentation by the Colonic Microflora
1.5.1 lntestinal Glycosidases
The human gastrointestinal tract contains a large number of glycosidases capable of hydrolyzing di- and oligosaccharides as well as polysaccharidases responsible for the hydrolysis of various polysaccharides [22,91]. Glycosidases are produced endogenously by mammalian cells as well as by bacteria in the colon. The composition of glycosidase activity in the gastrointestinal tract varies according to location (i.e., the stomach, the small intestine, and the colon). The glycosidases of the stomach and small intestine are bound to the cell membranes or excreted from the pancreas in the bile [92], whereas the glycosidase activity of the colon originates mainly from bacteria. Glycosidases produced by bacteria of the human colon are listed in Table 3. The glycosidases, which have the highest activity, are a-L-arabinofuranosidase, b-D-xylosidase, b-D-galactosidase, and b-D-glucosidases [91].
1.5.2 Intestinal Polysaccharidases
Living organisms cannot only synthesize biopolymers such as proteins, nucleic acids and polysaccharides, but are also capable of degrading them. The general mechanism of degradation of polymers in to small molecules employed by nature is a chemical one. Living organisms are capable of producing enzymes, which can attack biopolymers. The attack is usually specific with respect to both the enzyme/polymer couple and the site of attack at the polymer. Frequently, enzymes are designed according to their mode of action. Hydrolases, for instance, are enzymes catalyzing the hydrolysis of ester-, ether- or amide linkages. Proteolytic enzymes are called proteases and enzymes hydrolyzing polysaccharides are called glycosidases. There are three types of glucosidases that hydrolyze O-glucosyl, N-glucosyl or S-glucosyl bonds in polysaccharide chain. Table 4 describes the several enzymes together with their potential cleavage sites in different polysaccharides [93,94].
Polysaccharides are also called glycans; differ from each other in the identity of their recurring monosaccharide units, in the length of their chain, in the type of the O-glycosidic bonds linking the units, and in the degree of branching. Some polysaccharides serve as storage forms of monosaccharides, whereas other serves as structural elements in cell walls and connective tissues. On complete hydrolysis with acid or by the action of specific enzymes, polysaccharides yield monosaccharides or their derivatives. The polysaccharides can be divided in to several groups based on their source: microbial, plants, algae and sew weeds and animals (Table 5). Allowing indigenous microbes to assume some of the responsibility for carbohydrate breakdown presents a potential problem: How does the host derive nutritional benefit from the products of this breakdown? Monosaccharides released from carbohydrate polymers are converted in bacterial cells to pyruvate via glycolysis, resulting in net production of adenosine triphosphate (ATP) (Figure 5). Because, the distal intestinal lumen has a highly anaerobic environment, additional carbon and energy is extracted from pyruvate by microbial fermentation. Polysaccharides can be divided into starch and non-starch polysaccharides. Starch is the only polysaccharide that is degraded in the small intestine by the pancreatic enzyme a-amylase.
Some starches, however, are so-called resistant starches, which are stable and not digested until arrival in the large intestine [108]. Non-starch polysaccharides include plant cell wall components cellulose and hemicellulose, chemically related compounds such as the storage polysaccharides inulin and guar gum, seed mucilages such as ispaghula and plant gums, and exudates such as sterculia and karaya. In a study performed by Englyst and Cummings, it was found that the breakdown of non-starch polysaccharides in the stomach and small intestine was negligible [92].
Table 4. Several enzymes together with their potential cleavage sites in different polysaccharides93,94.
Enzymes |
Preferred cleavage sites |
Agarase |
Hydrolysis of 1,3-b-D-Galactosidic linkage in agarose |
µ-Amylase |
Hydrolysis of 1,4-µ-D-glucosidasidic linkages containing three or more 1,4- -linked D-glucose units |
b-Amylase |
Hydolysis of 1,4-µ-linked D-glucosidic linkages from nonreducing ends of chains |
k-Carageenase |
Hydrolysis of more 1,4-b-linkages between D-galactose-4-sulfate and 3,6-anhydro-D-galactose in carrageenans |
Cellulase |
Endohydrolysis of 1-4-b-D glucosidic linkages in cellulase |
Chitinase |
Random hydrolysis of N-acetyl-b-D-glucosaminide 1,4 b-linkages in chitin and chitodextrin |
Dextranase |
Endohydrolysis of 1,6-µ-D-glcosidic linkages in dextran |
µ-L-Fucosidase |
µ-L-Fucosidase+water yields an alcohol +L-fucose |
µ-Galactosidase |
Hydrolysis of terminal, nonreducing µ-D-galactosidase residues in µ-D-galactosides |
b-Galactosidase |
Hydrolysis Of Terminal, Nonreducing b-D-Galactosidase Residues In b-D-Galactosides |
µ-Glucosidase |
Hydrolysis of terminal, nonreducing 1,4-linked µ-D-glucose residues |
b-Glucosidase |
Hydrolysis of terminal, nonreducing b-D-glucose residues |
Hyaluronidase |
Random hydrolysis of 1,4 linkages between N-acetyl-b-D-glucosamine and D-glucuronate residues in hyaluronate and random hydrolysis of 1,3-linkages between N-acetyl-b-D-glucosamine and N-acetyl-D-glucosamine residues in hyluronate |
Inulinase |
Endohydrolysis of 2,1-b-D fructosidic linkages in inulin |
Isoamylase |
Hydrolysis of 1,6-µ-glucosidic branch linkages in glycogen, amylopectin, and their b-limit dextin |
Lysosyme |
Hydrolysis of 1,4 b-linkages between N-acetylmuramic acid and N-acetyl-D-glucosamine residues in petidoglycan and between N-acetyl-D-glucosamine residues in cyclodextrin |
Phosphorylase |
Cleavage of µ-D-glucose-L-phosphate from the non-reducing end of the amylose molecule. |
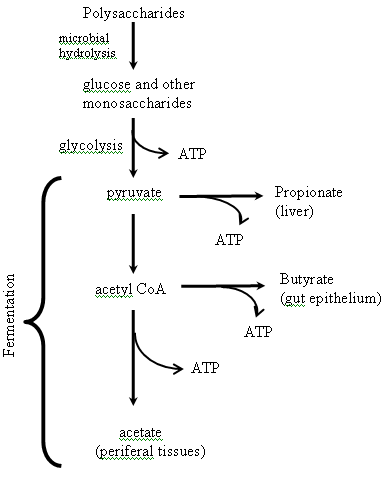
Figure 5: Overview of bacterial fermentation in the intestine. Monosaccharides released from carbohydrate polymers are converted in the bacterial cytoplasm to pyruvate via glycolysis, which results in net production of ATP. In the highly anaerobic environment of the distal intestine, additional carbon and energy is extracted from pyruvate by microbial fermentation. The predominant end-products of fermentation are acetate, propionate, and butyrate. To recover some of the nutritional value of polysaccharides degraded by gut microbes, mammalian hosts absorb and utilize these short chain fatty acid species.
Table 5. Main Structural Features of Polysaccharides Pursued/can be Pursued for Colon-Specific Drug Delivery.
Polysaccharide |
Main chains |
Side chains |
Source |
Comments |
Reference |
Amylose |
µ-1,4 D-glucose |
---- |
Storage |
Unbranched; one of |
95
|
Arabinogalactan |
b-1,4 and |
b-1,s and b-1,3 L-arabinose and D-galactose |
Plant cell walls |
Neutral pectin; |
96 |
Carrageenan |
Galactose residues which are sulfated and alternatively linked inµ-1,3 and b-1,4 linkage |
The 4 linked residues can be 3,6 anhydro-D-galactose and 3-linked residues are at least partly 4 sulfated. |
Sea weed extract |
---- |
97 |
Chitosan |
Deacetylated P-1,4 |
---- |
Shell of marine |
Deacetylated chitin; |
98 |
Chondroitin sulfate |
b-1,3 D-glucuronic |
---- |
Epithelial cells |
Mucopolysaccharide, |
97 |
| Dextran | µ-1,6 D-glucose | µ-1,3 D-glucose | Bacterial (Leuconostoc and Streptococcus) |
Used in medicine as plasma expander, degraded by Bacteroids |
99 |
Furcelleran |
Composed of D-galactose and 3,6-anhydro-D-galactose, with sulfate ester groups on both sugar components |
---- |
Sea weed extract (furcelleria fastigiata) |
---- |
99 |
Galactomannan |
µ-1,4 D-mannose |
µ-1,6 D-glucose |
Plant seed (endosperm of the seeds of cyamopsis tetragonolobus) |
Used in food industry |
100 |
Glucomannan |
random copolymer of (1-4) linked b-D mannose and b-D-glucose |
---- |
Amorphophallus konjac plant |
used as drug carrier, enzyme entrapment, reduces cholesterol level |
101 |
Gellan gum |
D-glucose, D-glucuronic acid and rhamnose in b-1,4 linkage |
---- |
Bacteria, extracellular |
---- |
102 |
Hyaluronic acid |
D- glucuronic acid and N-acetylglucosamine in b-1,3 and b-1,4 linkage, linear |
---- |
Human and animal (intercellular material in the space between skin, cartilage and muscle cells) |
---- |
103 |
Karaya gum |
Mixture of D-galactose, L-rhamnose and D-galactouronic acid |
The galctouronic acid units are the branching points of the molecule |
Plant (sterculia urens) |
---- |
104 |
Locust bean gum |
branched b-1,4-D-galactomannan units. |
---- |
Ceratonia |
it requires |
105 |
Scleroglucan |
(1→3)-linked b-D-glucopyranosyl units |
At every third unit it bears a single b-D-glucopyranosyl unit linked (1→6) |
Sclerotium (fungi) |
Used widely in industry, used for laxative, antitumor, antimicrobial effects |
106 |
Pullulan |
Maltotriose in µ-(1,4) |
Linkage connected by µ-1,6 linkage |
Fungi, extracellular (aurebasidium pullalans) |
---- |
107 |
Xylan |
b-1,4 D-xylose |
b-1,3 L-arabinose |
Plant cell walls |
The most abundant hemicellulose, degraded by Bacteroids, Bifidobacterium |
96 |
Bacteria in the colon obtain their main energy from the anaerobic fermentation of carbohydrates (i.e., residual starch or resistant starch and non-starch polysaccharides (dietary fiber)) resulting in the production of short-chain fatty acids (SCFA), also referred to as volatile fatty acids (VFA), and gases [109,110]. The three-major SCFA produced in the colon are acetate, propionate, and butyrate (Figure 5). In humans, the molar ratio of these three SCFA is approximately 70:20:10. Their aggregate concentration ranges from 70 to 120 mM [110-112]. However, these ratios and concentrations are influenced by diet. Conversion of pyruvate to any of these SCFA yields an additional molecule of ATP. Even after microbial extraction of ATP, SCFA production appears to represent 60–75% of the energy content of ingested carbohydrate [110].
In the cecum and right colon, high growth rates of bacteria exist and extensive fermentation takes place due to the high abundance of polysaccharides and other nutrients. This decreases from the right to the left colon, which in turn contains little bacterial growth and low fermentation because the nutrient flow in this region is low. Because SCFA are products of fermentation, their presence can be used as an indication of the fermentative activity in a specific region. Much of our understanding of the nutritional value of SCFA comes from studies of ruminants. The evolutionary success of ruminants in the wild is found on their ability to feed on cellulose-rich plants. These animals harbor a microbiota in their rumen that is responsible for digestion of carbohydrate polymers.
Some bacteria are able to degrade and ferment polysaccharides themselves due to their own polysaccharidases. The formed oligosaccharide fragments are then further fermented by saccharolytic bacteria not producing polysaccharidases by themselves. The most predominant SCFA produced are acetate, propionate, and butrate. Lactate and succinate are intermediates that can be further metabolized by saccharolytic bacteria forming the acetate, propionate, or butyrate SCFA [91]. It was found that 35 to 59% of carbon in polysaccharides would become SCFA, with a maximum of 70% [108]. The rate at which a given polysaccharides [91,113] degraded and the composition of the resulting product mixture is greatly dependent on the substrate as well as the enzymatic activity at that specific time [108]. Coupling these types of functional genomic analyses with enumeration studies of the gut microbiota should help in providing a molecular explanation for epidemiological data that suggest that dietary fiber reduces the incidence of colorectal cancer [114,115].
Bacterial polysaccharidases are induced by the presence of polysaccharides and are for the most part cell associated [91,113]. Only few polysaccharidases of bacterial origin have been isolated and characterized so far, and much polysaccharidase activity can probably be attributed to a mixture of enzymes (Table 6). The polysaccharide-degrading enzymes of the colon are produced by a variety of mainly anaerobic bacteria, of which bacteroides are the predominant species [108,116-119]. The enzyme activity and composition of the colonic microflora was in general found to be rather stable, as measured by the formation of end products, ammonia and SCFA, both inter- and intraindividually.
2. POLYSACCHARIDE CARRIERS
The polysaccharides naturally occurring in plant (eg., pectin, guar gum, inulin), animal (eg., chitosan, chondroitin sulfate), algal (eg., alginates), or microbial (eg., dextran) origin were studied for colon targeting. These are broken down by the colonic microflora to simple saccharides. Most of the polysaccharide-based delivery systems protect the bioactive from the hostile conditions of the upper GIT. Hydrolysis of the glycosidic linkages on arrival in the colon triggers the release of the entrapped bioactive. The main saccharolytic species responsible for this biodegradation are Bacteroides and Bifidobacteria. The colon-targeted delivery systems based on these polysaccharide systems with minimal chemical modifications will be discussed in detail.
Table 6. Bacterial Species Involved in the Degradation of Various Polysaccharides108,116-119.
Polysaccharide |
Bacterial species |
Cellulose |
Bacteroides |
Chondroitin sulfate |
Bacteroides |
Cyclodextrin |
Bacteroides |
Dextran |
Bacteroides |
Galactomannan (Guar gum) |
Bacteroides, ruminococci |
Gum arabic |
Bifidobacteria |
Mucin |
Bacteroides, bifidobacteria, ruminococci |
Non-cellulose p-glucans |
Bacteroides |
Pectin |
Bacteroides, bifidobacteria, eubacteria |
Starch |
Many (e.g., bacteroides, bifidobacteria) |
Xylan |
Bacteroides, bifidobacteria |
Glucomannan |
Bacteroids |
2.1 STARCH POLYSACCHARIDES
2.1.1 Starch
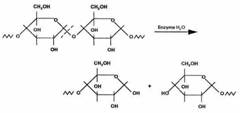
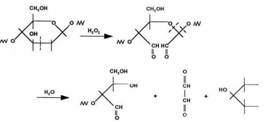
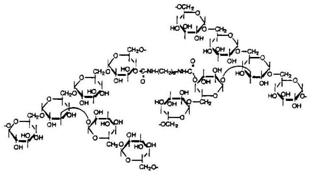
Starch is a polymer, which occurs widely in plants. In general, the linear polymer, amylose, makes up about 20wt % of the granule, and the branched polymer, amylopectin, the remainder. Amylose is crystalline and can have a number average molecular weight as high as 500000, but it is soluble in boiling water. Amylopectin is insoluble in boiling water, but in their use in foods, both fractions are readily hydrolyzed at the acetal link by enzymes. The a-1, 4-link in both components of starch is attacked by amylases (Figure 6) and the a-1, 6-link in amylopectin is attacked by glucosidases.
 Starch and its degrading products |
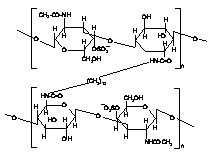 . .Crosslinked Chondroitin Sulfate with Diaminododecane |
 Amylose |
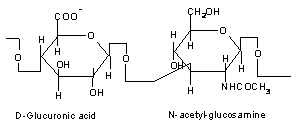 Hylauronic acid |
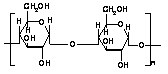 Cellulose and its degrading products |
 Chitosan |
 Pectin |
 Dextran |
 Inulin |
 Crosslinked Dextran with diisocyanate |
 Guar Gum |
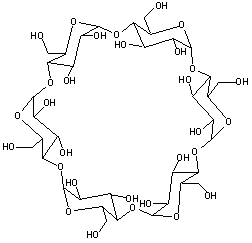 Cyclodextrins |
 Locust Bean Gum |
 Curdlan |
 Glucomannan |
|
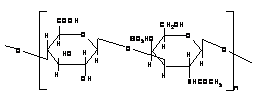 Chondroitin Sulfate |
Calcium Alginate
|
 Scleroglucan |
|
Starch has been evaluated for colon-targeted delivery as enteric-coated capsules [120]. The use of resistant starch was studied for the improvement of gut microflora and to improve clinical conditions related to inflammatory bowel diseases, immunostimulating activities, and protection from colon cancer. The resistant starch was modified chemically, preferably by etherification, esterification, or acidification. It may be hydroxypropylated, acetylated, octenyl succinylated, carboxymethylated, or succinylated. The enzymatic and/or physical modification by crystallization was also carried out [46]. The administration of probiotic bacteria with optionally modified resistant starch as a carrier and growth medium to alter the gastrointestinal tract microbial populations has resulted in a number of significant advantages, such as protection, of probiotics, and as a carrier to deliver economically and efficiently to specific sites. The enhancement of a resident microorganism population in a selected site of the GIT and suppression of an undesirable microbial pathogen are the other claims made by the usage of these formulations containing modified resistant starch and a probiotic. Other applications include reducing the incidence of colorectal cancers or colonic atrophy [121].
2.1.2 AMYLOSE
Amylose is a poly (1-4-α-D-glucopyranose) that consists of D-glucopyranose residues linked by α-(1-4) bonds (Figure 6). Those substances, present naturally in the diet, have the advantages of being safe, nontoxic, and easily available. These are resistant to pancreatic α-amylase, but are degraded by colonic bacterial enzyme [122]. Mixed films of amylose and ethyl cellulose as coatings have shown a great potential as colon delivery carriers [123]. Delayed release compositions comprising glassy amylose and an active compound were designed to permit the release when the composition reaches the large intestine. The release of the active compound was reported to be delayed in an aqueous environment of pH 1-9 at 37oC. The release was triggered when exposed to an enzyme capable of clearing the amylase. The delivery system can be made into a powder or monolithic form. This composition is useful in the diagnosis and therapy of diseases of the colon. The above composition may also be applied for delivery of anti-arthritic drugs and also for pesticidal delivery [124].
Milojevic et al. [125] prepared and evaluated in vitro potential of amylose-ethocel coating system for colon-targeted delivery. Aqueous dispersions were prepared using amylose and ethocel, which was used to coat 5-ASA pellets to make them resistant to gastric and small intestinal enzymes. Amylose and Ethocel in coating ratio of 1:4 was optimum with least drug release in stomach and intestinal fluids. In vitro release of 5-ASA from coated pellets with amylose and EC (1:4) was retarded in simulated gastric and small intestinal fluid over a period of 12 hr. It was fermented and drug was released in 4 hr in simulated colonic environment containing mixed fecal bacteria of human origin. The potential of amylose in novel colon targeted drug delivery has been investigated using glucose pellets prepared by extrusion and spheronization. Various glucose formulations were studied in which glucose and Avicel® PH101 (microcrystalline cellulose) remained the same while the binding agent, used to retard glucose release, was changed. The dissolution media (400 mL at 37 ± 0.5oC) used were water, hydrochloric acid (pH 1.2 for 3 hr), and 0.2 M phosphate buffer (pH 7.2 for 9 hr) with simulated gastric and intestinal conditions, respectively. Pellet cores containing 20% glycerol monostearate, 50% glucose, and 30% Avicel® PH101 coated with amylose- Ethocel® (1:4 w/w) exhibited gastric and small intestinal resistance but were susceptible to bacterial enzymatic attack in simulated colonic conditions. In vitro fermentation studies were carried out in a batch fermentor inoculated using fresh feces homogenized in anaerobic 0.1 mL/L sodium phosphate buffer (pH 7.0). Carbon dioxide and volatile fatty acids, end-products of colonic bacterial fermentation, were measured over a subsequent 48 hr period, which showed an early release of glucose from coated pellets.
A mixture of amylose and Ethocel (1:4) has been developed and (13C) glucose used as surrogate for drug delivery. Eight healthy subjects were given amylose-Ethocel-coated pellets containing 300 mg (13C) glucose together with a capsule that contained amberlite resin labeled with 99mTc to act as a transit marker and outline for the anatomy of the gut. According to γ -scintigraphy, after 7.1 hr all the material was in the colon. Breath 13CO2 data were analyzed by the cusum technique and with total 13CO2 recovery at 25 hr that was found to be 37.5% [123]. Epichlorhydrin-treated crosslinked amylose was introduced as a matrix for controlled release of theophylline [126]. Amylose ethylcellulose film coating has been investigated for colon targeted drug delivery [127]. Drug release rate from the films was governed by the concentration of amylose and EC. Drug release rate was inversely proportional to the thickness of the coating and also influenced by the amount of amylose present in the film. These films were shown to be degraded by bacterial enzymes in simulated colonic environment.
2.1.3 CELLULOSE
Many polymer researchers are of the opinion that polymer chemistry had its origins with the characterization of cellulose. Cellulose was isolated for the first time some 150 years ago. Cellulose differs in some respects from other polysaccharides produced by plants, the molecular chain being very long and consisting of one repeating unit (cellobiose). Naturally, it occurs in a crystalline state. From the cell walls, cellulose is isolated in microfibrils by chemical extraction. In all forms, cellulose is a very highly crystalline, high molecular weight polymer, which is infusible and insoluble in all but the most aggressive, hydrogen bond breaking solvents such as N-methylmorpholine-N-oxide. Because of its infusibility and insolubility, cellulose is usually converted into derivatives to make it more processable. Some fungi can secrete enzymes that catalyze oxidation reactions of either cellulose itself or the lower molecular weight oligomers produced from the enzymatic hydrolysis of cellulose. Of these, the peroxidases can provide hydrogen peroxide for free radical attack on the C2–C3 positions of cellulose to form ‘aldehyde' cellulose, which is very reactive and can hydrolyze to form lower molecular weight fragments (Figure 6) while other oxidative enzymes can oxidize glucose and related oligomers to glucuronic acids. Bacteria also secrete both endo- and exoenzymes, some of which form complexes that act jointly in degrading cellulose to form carbohydrate nutrients, which is utilized by the microorganisms for survival [128, 129].
In anaerobic environments such as in colon, a variety of final products are formed, including CO2 hydrogen, methane, hydrogen sulphide and ammonia. CO2 can be formed by oxidative reactions, which utilize inorganic compounds, such as sulphate and nitrate ions, in the environment as oxidizing agents. Hydrogen produced by some anaerobic bacteria can be utilized by autotrophic bacteria to reduce oxidized compounds and CO2 to form either acetic acid or methane.
2.2 POLYSACCHARIDES FROM PLANT SOURCE
2.2.1 PECTINS
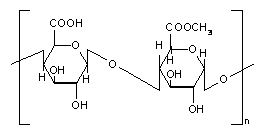
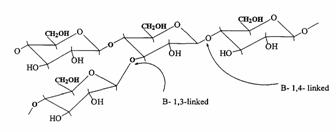
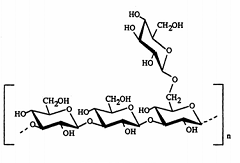
Pectins are nonstarch linear polysaccharides that consist of α-1,4 D-galacturonic acid and 1,2 D-rhamnose with D-galactose and D-arabinose side chains having average molecular weights between 50,000 to 150,000 (Figure 6). Pectin tends to produce lower viscosities than other plant gums. It is refractory to host gastric and small intestinal enzymes but is almost completely degraded by the colonic bacterial enzymes to produce a series of soluble oligalactorunates [130,131] Depending on the plant source and preparation; they contain varying degrees of methyl ester substituents [132]. Pectin is highly soluble in water, which put hurdles in the development of colon targeted drug delivery systems. If used alone it swells when it comes in contact with aqueous fluids of GIT and causes the release of the entrapped drug through the diffusion. This problem can be manipulated through choice of pectin type or the presence of additives [133,134]. Another way to alleviate this problem is by the use of hydrophobic polymers, e.g., ethylcellulose. It restricts the entry of water and consequently swelling of polymer. Moreover, a better shielding effect can be obtained by reducing the solubility of pectin by forming its calcium salt, calcium pectinate. The degree of methylation (DM) has an essential influence on the properties of pectin, especially on its solubility and its requirements for gelation, which are directly derived from the solubility. The DM of 50% divides commercial pectins into high methoxy pectins and low methoxy pectins. These two groups of pectin are gelled by different mechanisms. High methoxy pectins require a minimum amount of soluble solids and a pH within a moderate range around 3 to form gels. Low methoxy pectins require the presence of a controlled amount of calcium ions for gelation and need neither sugar nor acid. Low methoxy pectin gelation resembles the behavior of alginate. Drug delivery systems utilizing pectin is discussed by various researchers [7,135-137] therefore, it will not be discussed in detail any further. But for readers I am citing few references on pectin mediated colon specific drug delivery [60,61,138-151].
2.2.2 INULIN
Inulin is a naturally occurring storage polysaccharide found in many plants such as onion, garlic, artichoke, and chicory. Chemically, it belongs to the glucofructans and consists of a mixture of oligomers and polymers containing 2 to 60 (or more) β-2-1 linked D-fructose molecules (Figure 6). Most of these fructose chains have a glucose unit as the initial moiety. It is not hydrolyzed by the endogenous secretions of the human digestive tract [152]. However, bacteria harboring in the colon and more specifically Bifidobacteria are able to ferment inulin [153-155]. The inulin has been incorporated into Eudragit RS films for preparation of mixed films that resisted degradation in the upper GIT but digested in human fecal medium by the action of Bifidobacteria and Bacteroids [156]. Various inulin hydrogels have been developed that serve as potential carriers for the introduction of drugs into the colon [157-160]. Vinyl groups were introduced in inulin chains to form hydrogels by free radical polymerization. Inulin was reacted with glycidyl methacrylated in N,N-dimethylformamide in the presence of 4-dimethylaminopyridine as catalyst. 1H and 13C NMR spectroscopy were used for the characterization of the obtained reaction product and revealed the conversion of the incorporated vinyl groups into covalent crosslinks upon free radical polymerization of aqueous solutions of the derivatized inulin [157]. Maris et al. [160] synthesized and characterized new hydrogel systems composed of methacrylated inulin, copolymerized with the aromatic azo agent BMAAB and HEMA or MA. The gels were assessed by studying the influence of various parameters on the dynamic and equilibrium degree of swelling. Inulin hydrogels have been developed and characterized for dynamic and equilibrium swelling properties and in vitro degradation study [158,159]. The rate of water transport into the inulin hydrogels was quite high (mean swelling time <1.2 hr) and the hydrogels exhibited anomalous dynamic swelling behavior. The equilibrium swelling of the hydrogels was influenced by the degree of substitution and feed concentration of methacrylated inulin, by the initiator concentration, by the ionic strength, and by an acidic pH of the swelling solvent. The enzymatic digestibility of the prepared inulin hydrogels was assessed by performing an in vitro study using an inulinase preparation derived from Aspergillus Niger. The amount of fructose liberated from the inulin hydrogels by the action of inulinase was quantified using the anthrone method. The equilibrium swelling ratio as well as the mechanical strength of the hydrogels was studied before and after incubation in inulinase solutions. The data obtained by these different methods indicate that enzymatic digestion of the inulin hydrogels appeared to be enhanced by a prolonged degradation time, a higher inulinase concentration, and a lower degree of substitution and feed concentration of the hydrogel polymer. The inulin hydrogels exhibited an increase in equilibrium swelling after degradation compared with the swelling before degradation, suggesting that inulinase enzymes diffuse into the inulin hydrogel networks causing bulk degradation. Stubbe et al. [161] developed azo-containing polysaccharide gels more specifically azo-inulin and azo-dextran gels. Compared with azo-hydrogels that can only be degraded by reduction of the azo groups, the goal of the study was to evaluate whether, in vitro, azo-polysaccharide gels could be degraded through both reduction of the azo-groups in the crosslinks as well as enzymatic breakdown of the polysaccharide backbone. The azo-polysaccharide gels were synthesized by radical crosslinking of a mixture of methacrylated inulin or methacrylated dextran and BMAAB and were characterized by dynamic mechanical analysis and swelling measurements. Azo-dextran gels could be obtained from methacrylated dextran having low degree of substitution but not from lowly substituted methacrylated inulin. Increasing the amount of BMAAB resulted in denser azoinulin and azo-dextran networks. Compared with their swelling in dimethylformamide, all azo-dextran gels became more swollen in water while azo-inulin gels shrank upon exposure to water, indicating a more hydrophobic character of the azoinulin gels. Breakdown of the inulin and dextran chains in the azo-polysaccharide gels by inulinase and dextranase, respectively, was observed. However, the degradation of azo-dextran gels by dextranase seemed more pronounced than the degradation of the azo-inulin gels by inulinase. In rat caecal content medium, reduction of the azo function in azo-inulin gels was not observed. This may be attributed to a low partitioning of nicotinamide-adenine dinucleotide phosphate in the gels.
2.2.3 GUAR GUM
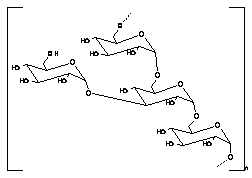
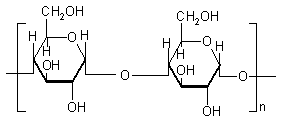
Guar gum, obtained from the ground endosperms of Cyamposis tetragonolobus, consists of chiefly high molecular weight hydrocolloidal polysaccharide, composed of galactan and mannan units combined through glycosidic linkages and shows degradation in the large intestine due the presence of microbial enzymes [162-165]. The structure of guar gum is a linear chain of β-D-mannopyranosyl units linked (1→4) with single member α-D-galactopyranosyl units occurring as side branches (Figure 6) [166]. It contains about 80% galactomannan, 12% water, 5% protein, 2% acid soluble ash, and 0.7% fat. Guar gum has a molecular weight of approximately 1 million, giving it a high viscosity in solution. This galactomannan is soluble in cold water, hydrating quickly to produce viscous pseudoplastic solutions that although shear-thinning generally have greater low-shear viscosity than other hydrocolloids [167]. This gelling property retards release of the drug from the dosage form, and it is susceptible to degradation in the colonic environment. Homogenized and diluted feces from human source were incubated with the guar gum to investigate the degradation of polysaccharide by intestinal microflora. It produced a rapid decrease in viscosity and fall in pH while no such results were observed when it was incubated with autoclaved fecal homogenates [168]. Guar gum is being extensively studied by many researchers for colon targeting [7,135,136,169-178] therefore; it will not be discussed in detail any further.
2.2.4 LOCUST BEAN GUM
It is also called carob gum, as it is derived from carob (Ceratonia siliqua) seeds. Locust bean gum has an irregularly shaped molecule with branched β-1,4-D-galactomannan units (Figure 6). This neutral polymer is only slightly soluble in cold water; it requires heat to achieve full hydration and maximum viscosity. Cross-linked galactomannan however led to water-insoluble film forming product-showing degradation in colonic microflora [179]. The colon-specific drug delivery systems based on polysaccharides, locust bean gum and chitosan in the ratio of 2:3, 3:2 and 4:1, were evaluated using in vitro and in vivo methods [180]. Core tablets containing mesalazine with average weight of 80 mg were prepared by compressing the materials using 6-mm round, flat, and plain punches on a single station tablet machine. The formulated core tablets were compression coated with different quantities of locust bean gum and chitosan. The in vitro studies in pH 6.8 phosphate buffer containing 2%w/v rat caecal contents showed that the cumulative percentage release of mesalazine after 26 hr was 31.25 ± 0.56, 46.25 ± 0.96, and 97.5 ± 0.26, respectively. The in vivo studies conducted in nine healthy male human volunteers for the various formulations showed that the drug release was initiated only after 5 hr, which is the transit time of small intestine. In vitro drug release studies and in vivo studies revealed that the locust bean gum and chitosan as a coating material applied over the core tablet was capable of protecting the drug from being released in the physiological environment of stomach and small intestine and was susceptible to colonic bacterial enzymatic actions with resultant drug release in the colon. Hirsch et al. [181] carried out dissolution studies on theophylline tablets coated with crosslinked locust bean gum and showed the mechanical instability of these coatings in the dissolution media, thereby suggesting the non-suitability of such films as colon carriers.
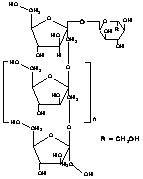
2.2.5 GLUCOMANNAN
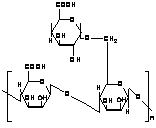
Konjac Glucomannan (KGM) is a high-molecular weight water-soluble non-ionic glucomannan extracted from tubers of the Amorphophallus konjac plant. Konjac glucomannan (KGM) is a linear random copolymer of (1→4) linked b-D mannose and b-D-glucose. It has mannose and glucose units in a molar ratio of 1.6:1 with a low degree of acetyl groups (approximately 1 acetyl group per 17 residues) at the C-6 position [182-183]. The degree of solubility is controlled by the presence of acetyl groups. Figure 6 shows the chemical structure of KGM as proposed by Maeda et al. [101].
It is similar to pectin, which is not hydrolyzed by digestive enzyme in human being and is considered as an indigestible dietary fiber that has received recognition for reducing the risk of developing diabetes and heart disease. A diet rich in high-viscosity KGM is generally recommended for improved glucose and lipid profile control, suggesting a therapeutic potential in the treatment of the insulin-resistance syndrome [184]. KGM could be hydrolyzed by b-mannanase to manufacture manno-oligosaccharides, which play important roles in biological systems [185]. In addition, KGM could form strong, elastic, heat stable gels when heated with mild alkali [186], and has also been used as drug carrier [187], food preservative [188,189] and enzyme entrapment [190]. However, the encapsulation yield of protease remained only about 50%, and KGM loses the above physiological action after alkali treatment and heating [190]. Konjac glucomannan has the ability to lower blood cholesterol and sugar level, help with weight loss, promote intestinal activity and immune function etc. At the same time, this polysaccharide can be prepared into various derivatives easily because of its good biocompatibility and biodegradable activity. KGM may be very promising polysaccharide with respect to colon specific drug delivery systems but only limited drug delivery systems have been tried till date [191-193].
Konjac glucomannan is a kind of polysaccharide with wide uses. Although they have been utilized and investigated for so many years, konjac glucomannan and its derivatives still need to be well investigated compared with other polysaccharides such as cellulose and starch etc. There is a still lot of work to be done for understanding of this interesting polysaccharide and more promising drug delivery systems and products developed from it.
2.2.6 KHAYA AND ALBIZIA GUMS
Two naturally occurring plant gums, khaya and albizia gums, are also under evaluation for drug targeting to the colon. Khaya gum is a polysaccharide obtained from the incised trunk of the tree Khaya grandifoliola (family Meliaceae). It is known to contain highly branched polysaccharides consisting of D-galactose, L-rhamnose, D-galacturonic acid and 4-O-methyl-D-glucoronic acid [194]. Khaya gum has been shown to be useful as a binding agent in tablet formulations. Khaya gum is a hydrophilic polymer and has been shown to possess emulsifying properties comparable with acacia gum. The fact that the gum is naturally available, inexpensive and non-toxic has also fostered the interest in developing the gum for pharmaceutical use. Further work has also shown its potential as a directly compressible matrix system in the formulation of controlled release tablets [195].
Albizia gum is obtained from the incised trunk of the tree Albizia zygia (DC) J. F. Macbr, family Leguminosae and is shaped like round elongated tears of variable color ranging from yellow to dark brown. It consists of b-1– 3-linked D-galactose units with some ß1-6-linked D-galactose units. The genus Albizzia containing some twenty-six species is a member of the Mimosacez, a family which also includes the gum-bearing genera Acacia and Prosopis. Only two species of Albizia, A. zygia and A. sassa, are however, known to produce gum. Albizia gum has been investigated as a possible substitute for gum Arabic as a natural emulsifier for food and pharmaceuticals [196,197].
These gums were tried as coating materials in compression-coated tablets, which degraded, by the colonic microflora, thereby releasing the drug. Recent studies on the binding properties of albizia gum have also shown that the gum produces tablets with greater mechanical properties and longer disintegration and dissolution times when compared with gelatin BP.
Odeku and Fell [195] evaluated Khaya gum as a controlled release agent in modified release matrices in comparison with hydroxypropylmethylcellulose (HPMC) using paracetamol (water soluble) and indomethacin (water insoluble) as model drugs. Tablets were produced by direct compression and the in-vitro drug release was assessed in conditions mimicking the gastrointestinal system. Khaya gum matrices provided a controlled release of paracetamol for up to 5 h. The release of paracetamol from khaya gum matrices followed time-independent kinetics and release rates were dependent on the concentration of the drug present in the matrix. Researchers found that Indometacin exhibited a lag time in excess of 2 h, due to its insolubility at low pH, before the zero-order release was observed. Thus khaya gum matrices could be useful in the formulation of sustained release tablets for up to 5 h and the appropriate combination of khaya gum and HPMC could be used to provide a time-independent release for longer periods.
Odeku and Fell [198] used khaya gum, albizia gum and a mixture of both as compression coats and evaluated their ability to prevent drug release under conditions mimicking the mouth to colon transit time using indometacin (a water insoluble drug) and paracetamol (a water soluble drug) as model drugs. The susceptibility of the gums to undergo biodegradation in the colon was assessed by conducting drug release studies in the presence of rat caecal contents in phosphate-buffered saline (PBS) at pH 6.8. While Odeku and Fell observed in their work that after 5 h, 21% of indomethacin and 19% of paracetamol were released for tablets coated with khaya gum, while tablets coated with albizia gum released less than 4% of indomethacin and 6% paracetamol. Tablets containing the khaya/albizia mixture released less than 5% of indomethacin and 9% of paracetamol. This suggests that the khaya and albizia gums can effectively control the release of the drugs in the physiological environment of the stomach and small intestine.
The results demonstrate that khaya and albizia gums have great potential for drug targeting to the colon. But till date not many drug delivery systems have been investigated utilizing these gums for targeting to colon.
2.3 POLYSACCHARIDES FROM ANIMAL SOURCE
2.3.1 CHONDROITIN SULFATE
Chondroitin sulfate is a soluble mucopolysaccharide [199,200] that is used as a substrate by the bacteroid inhabitants of the large intestine mainly by Bacteroides thetaiotaomicron and B. ovatus (Figure 6) [201]. Periplasmic enzymes are probably responsible for chondroitin breakdown apparently an outer membrane receptor binds chondroitin sulfate and brings it in contact with enzymes like chondroitin-sulfate-lyase [81]. Chondroitin sulfate could be used as carrier for colon targeted delivery of bioactive agents. However, use would depend on its persistence as a solid dosage form in the physiological environment of the stomach and small intestine. Since natural chondroitin sulfate is readily water-soluble, it might not protect its load successfully. However, crosslinked chondroitin sulfate would be less hydrophilic and thus would provide a better shield. Chondroitin sulphate was crosslinked with 1,12 diaminododecane using dicyclohexylcarbodiimide as a catalyst and formulated in a matrix with indomethacin as a drug marker (Figure 6). The indomethacin release kinetics from the various formulations was analyzed in PBS with and without rat caecal content at 37oC under carbon dioxide atmosphere. It was concluded that the release of indomethacin depended upon the biodegradation action of the caecal content [202,203]. The extent of crosslinking was assessed by the amount of methylene blue that adsorbed as a result of cation exchange. The crosslinked chondroitin dry powder was sieved and mixed with indomethacin for the preparation of matrix tablets. The indomethacin release kinetics from the various formulations was analyzed in PBS with and without rat caecal content at 37◦C under a carbon dioxide environment. In separate experiments, the caecal content was sonicated to cause the lysis of the bacterial cell membranes. The drug release profiles indicated a constant rate of biodegradation in the presence of rat caecal content, and diffusion-driven release from the matrix’s surface area in PBS control. Prolonged incubation in PBS with rat caecal content increased drug release and after 2 hr the released indomethacin levels were significantly higher than those in the PBS controls. The maximal cumulative percent drug release values for the PBS controls were 30.1 ± 10.0, 19.7 ± 15.0, and 9.0 ± 4.1 for the three levels of crosslinkage of the chondroitin sulfate carriers, respectively, whereas those for the caecal content media were 71.0 ± 19.0, 48.0 ± 35.0, and 22.5 ± 7.9, respectively. The linear correlation between the degree of crosslinking and the amount of drug released in the presence of caecal content suggests that drug release in the colon can be controlled by adjusting the relative amounts of the variously crosslinked chondroitin sulfate formulations in the matrices. Sintov et al. (2004) crosslinked chondroitin sulphate with 1,12-diaminododecane to give a series of crosslinked products with reduced water solubility. Indomethacin tablets were prepared using two types of crosslinked polymers: very low water soluble and relatively high water soluble and analyzed for their water uptake and drug release characteristics.
2.3.2 HYALURONIC ACID
Hyaluronic acid (HA) is a naturally occurring biopolymer, which serves important biological functions in bacteria and higher animals including humans. Naturally occurring HA may be found in the tissue of higher animals, in particular as intercellular space filler. It is found in greatest concentrations in the vitreous humour of the eye and in the synovial fluid of articular joints [205].
Since its discovery in human tissue, HA and its derivatives has been largely studied and applied in the biomedical arena. Its high level of biocompatibility has accentuated the appeal of this polymer. It has been used in viscosurgery to allow surgeons to safely create space between tissues. As a microcapsule it can be used for targeted drug delivery [103].
The utility of this biopolymer is derived from a remarkably simple construct. HA is comprised of linear, unbranching, polyanionic disaccharide units consisting of glucuronic acid (GlcUA) an N-acetyl glucosamine (GlcNAc) joined alternately by b-1-3 and b-1-4 glycosidic bonds (Figure 6). It is a member of the glycosaminoglycan family which includes chondroitin sulphate, dermatin sulphate and heparan sulphate. Unlike other members of this family, it is not found covalently bound to proteins. When incorporated into a neutral aqueous solution hydrogen bond formation occurs between water molecules and adjacent carboxyl and N-acetyl groups. This imparts a conformational stiffness to the polymer, which limits its flexibility. The hydrogen bond formation results in the unique water-binding and retention capacity of the polymer. It also follows that the water binding capacity is directly related to the molecular weight of the molecule. Up to six liters of water may be bound per gram of HA. HA solutions are characteristically viscoelastic and pseudoplastic. This rheology is found even in very dilute solutions of the polymer where very viscous gels are formed. The viscoelastic property of HA solutions which is important in its use as a biomaterial is controlled by the concentration and molecular weight of the HA chains. The molecular weight of HA from different sources is polydisperse and highly variable ranging from 104 to 107 Da. The extrusion of HA through the cell membrane as it is produced permits unconstrained polymer elongation and hence a very high molecular weight molecule.
Colon cancer and inflammatory bowel disease are two devastating and relatively common diseases of the large intestine. Colon cancer, a malignant disorder that affects an older population typically metastasizes to other organ systems. Conversely, inflammatory bowel disease, a benign condition of younger individuals, is characterized by inflammation of the gut as well as of some extra-intestinal sites. The metastatic potential of certain colon carcinomas correlates with the expression of the hyaluronan receptor CD44, which circumstantially links hyaluronan with tumor progression. CD44 is frequently overexpressed on many colon carcinomas, as are some of its isoforms. The variant CD44v6 is most commonly expressed in advanced stages of tumor development and strongly correlates to colorectal tumor-related deaths [206]. Recent evidence also suggests that stromal cells of the intestine synthesize hyaluronan in response to factors produced by adenocarcinomas [207]. Rudzki et al. [208] reported that CD44 transcripts are strongly over expressed in tumors compared to normal colon. Both neoplastic and normal colon samples exhibited the same species of CD44 transcript representing standard and variant isoforms. Seventy-five percent of neoplasms contained foci of CD44-positive tumor cells, whereas in normal colon the epithelial immunoreactivity was confined to the crypt base.
Hyaluronan is moderately abundant in colonic tissue and plays an important role in tissue osmosis. The origin of inflammatory bowel disease appears to be multifactorial; environmental and microbiological factors initiate and perpetuate an immune response in the intestine of genetically susceptible individuals. Some evidence suggests that hyaluronan may play a pivotal role in this inflammatory disorder. Increased hyaluronan levels have been measured in the intestine of patients with inflammatory bowel disease. Also, compared with other inflammatory disorders, greater levels of CD44 are observed throughout the inflamed colon tissue of patients with ulcerative colitis, a specific type of inflammatory bowel disease, and in particular increased amounts of CD44v6 and CD44v3 are expressed by the epithelial cells. Interestingly, the extra-intestinal manifestations of inflammatory bowel disease, which are exhibited in 30-50% of patients, occur in hyaluronan-rich tissues (e.g., eye, joint, and skin). Immune phenomena linked to the etiopathogenesis of inflammatory bowel disease have also been extensively investigated, and findings have underscored the differences between the responses of normal and affected individuals. For example, it is well known that the distribution of specific cytokine-producing lymphocytes differs between normal persons and patients with Crohn's disease or ulcerative colitis. However, much less is known about how microbial agents affect the disease process. Speculations that viruses may be involved in the pathogenesis of inflammatory bowel disease have been advanced for some time because of the clinical association of respiratory virus infections with subsequent disease flares. With inflammatory bowel disease, the mucosal immune cell population increases dramatically, and the infiltrate is predominantly comprised of mononuclear leukocytes. Furthermore, the muscularis mucosae cell layer thickens to nearly 300 times its normal depth secondary to smooth muscle cell hyperplasia and extracellular matrix deposition. These features suggest that interactions between recruited leukocytes and mesenchymal smooth muscle cells are important in the development and propagation of inflammatory bowel disease.
HA is a kind of biopolymer with wide uses. Although they have been utilized and investigated for so many years, HA still needs to be well investigated compared with other polysaccharides such as chondriotin sulphate etc. There is a still lot of work to be done for understanding of this interesting biopolysaccharide and more promising drug delivery systems and products for colon specific drug delivery.
2.3.3 CHITOSAN
Chitosan is a functional linear polymer derived from chitin, the most abundant natural polysaccharide next to cellulose, and it is not digested in the upper part of the GIT by human digestive enzymes [62,209,210]. Chitosan is a copolymer consisting of 2-amino-2-deoxy- D-glucose units linked with β-(1→4) bonds (Figure 6). It should be susceptible to glycosidic hydrolysis by microbial enzymes in the colon because it possesses glycosidic linkages similar to those of other enzymatically-depolymerized polysaccharides. Drug delivery systems utilizing chitosan is discussed by various researchers [136,211-226] therefore, it will not be discussed in detail any further.
2.4 POLYSACCHARIDES FROM BACTERIAL SOURCE
2.4.1 DEXTRANS
This class of polysaccharide consists of linear chains of α-D glucose molecules, 95% of the chains consists of 1:6-α-linked linear glucose units while the side chains consist of 1:3-α-linked moieties (Figure 6). They are obtained from microorganisms of the family of Lactobacillus (Leuconostoc mesenteroides). Dextrans are colloidal, hydrophilic, and water-soluble substances, inert in biological system, and do not affect cell viability. Dextranases are the enzymes that hydrolyze glycosidic linkages of the dextrans. Anaerobic Gram-negative intestinal bacteria show Dextranase activity of the colon especially by the Bacteroides [117]. Dextran also has been found to be degraded in human feces due to bacterial action [227]. Dextrans of various molecular weights have been used as drug carriers. Various dextran ester prodrugs have been prepared and evaluated for their efficacy to deliver the drug to the target organ, i.e., colon. Harboe et al. [228,229] synthesized dextran ester prodrug. The bioavailability of naproxen after oral administration of aqueous solutions of a dextran T-70-naproxen ester was compared with that of an oral solution of an equivalent dose of naproxen in pigs. The bioavailability was close to 100% in comparison to the oral dose of parent drug, and drug activation took place in the lower part of the GIT. It was established from several features of the prodrug that naproxen was released from the prodrug prior to systemic absorption and that drug activation involved the action of one or more enzyme systems located in the GIT. In rabbits, the plasma concentration-time curves for the conjugate were characterized by an initial lag time of about 2–3 hr, whereas naproxen was detected in plasma immediately after per oral administration of the drug compound per se. The distribution of the prodrug along the GIT at various times after conjugate administration was assessed by HPLC analysis of conjugated and free naproxen in various segments of the GIT. From these experiments, drug regeneration was effective in the bowel below the ileum. Dextran ester prodrugs of ketoprofen and naproxen were prepared using dextran with molecular weight 10,000-50,0000 which showed degradation in the colon. In vitro release revealed that release of naproxen from prodrug was several folds higher in caecum homogenates than in control medium or homogenates of the small intestine of pig [27,230,231]. The side effects of steroid therapy, which are used in the treatment of chronic colitis, may be decreased by selectively delivering drug to the colon [211,232]. One potential way of achieving this is to attach the drug to dextran, a polar macromolecule that prevents drug absorption in the small intestine. Previous reports on colon-specific delivery using dextran prodrugs have focused on non-steroidal anti-inflammatory agents, which are directly coupled to dextran using the carboxylic acid groups of the drugs [228,233]. Experiments with these conjugates have demonstrated that dextranases and esterases in the colon degrade the conjugate and liberate drug at this site [230,234]. Glucocorticoids-dextran ester prodrugs have been prepared and proved efficacious in delivering drugs to colon [28,235,236]. Methylprednisolone and dexamethasone were attached to dextran with molecular weight 72,600 by a succinate linker and, in addition, dexamethasone was attached by glutaric acid to investigate the effect of linker molecules on hydrolysis kinetics. The kinetics of degradation of the hemiester and corresponding dextran conjugates were measured as a function of pH and temperature. Intermolecular migration of the linker molecule from the 21- to the 17-position on the glucocorticoid occurred in all three hemiester, although to a greater extent in methylprednisolonehemiester. The dextran conjugates were also incubated at 37oC, pH 6.8, and the chemical degradation half-lives was determined. Dextran hydrogels have been shown to be the promising carrier for the delivery of drugs to colon. Brondsted, Hovgaard, and Simonsen [237,238] prepared dextran hydrogels utilizing diidocyanate as crosslinker, which were shown to be degradable in vitro and in vivo in rat caecum (Figure 6). Simonsen et al. [239] reported the degradability of dextran hydrogels in the human colonic fermentation model; however, the fermentation of gels started after 24 hr, which would cause delay in drug release. Hovgaard and Brondsted [240] have studied the suitability of dextran hydrogels for colon-targeted delivery of hydrocortisone. The hydrogels were characterized by equilibrium degree of swelling and mechanical strength, and the degradation of hydrogels was studied in vitro using dextranase, in vivo in rats, and in human fermentation model. They found that by changing the chemical composition of the hydrogels, it is possible to control the equilibrium degree of swelling, mechanical strength, and biodegradability. The dextran hydrogels were degraded in vivo in the caecum of rats but not the stomach. Furthermore, the dextran hydrogels were degraded in a human colonic fermentation model, indicating that dextranses are indeed present in human colonic contents. Finally, release of hydrocortisone from the hydrogels was found to depend on the presence of dextranases in the release medium. pH-sensitive dextran hydrogels were prepared by activation of dextran (T-70) with 4-nitrophenyl chloroformate, followed by conjugation of the activated dextran with 4-aminobutyric acid and crosslinking with 1,10-diaminodecane. The crosslinking efficiencies determined by mechanical measurements were in the range of 52–63%. Incorporation of carboxylpropyl groups in dextran hydrogels led to a higher equilibrium and faster swelling under high pH conditions. The swelling reversibility of hydrogels was also observed after repeated changes in buffers between pH 2.0 and 7.4. The slow rates of swelling and deswelling in response to changes in pH were attributed to the hydrophilic nature of dextran and formation of hydrogen bonds between the hydroxyl groups of dextran with water molecules. The pronounced effect of carboxylic acid content on degradation of hydrogels was observed after 4 hr incubation with dextranase, and the influence significantly decreased after exposure to the enzyme for 8 hr. The mechanism of bulk degradation of hydrogels under high swelling extent was substantiated using Coomassie blue protein assay. The release rate of bovine serum albumin from hydrogels was primarily determined by the swelling extent. The release rate was further enhanced by addition of dextranase in buffer solutions [241]. Bauer and Kesselhut [179] synthesized dextran fatty acid ester and showed that lauroyl dextran esters with molecular weight of approximately 250,000 and degree of substitution ranging from 0.11 to 0.3 were suitable for colon targeted drug delivery as film coatings. In vitro studies with lauroyl dextrans esters bearing theophylline were carried out and Hirsch et al. [242] showed that addition of dextranase accelerated the drug release. However, further studies negated the utility of dispersion of lauroyl dextrans as coating material as they did not exhibit ideal zero-order dissolution before and quick disintegration after enzyme addition [181]. The dextran capsules appear to be excellent candidates for colon-specific drug delivery of almost any drug molecules as the polymer composition governs the specific release pattern [243]. A reaction mixture containing dextran, magnesium chloride, glutaraldehyde, and polyethyleneglycol 400 in water was applied onto molding pins of nylon producing capsule caps and bodies. The capsule materials were characterized by measuring the mechanical strength in compression and equilibrium degree of swelling. Based on these results, an optimal composition for the capsule material was selected. The dextran capsules were loaded with hydrocortisone and subsequently drug release was studied. The release was found to be about 10% during the initial 3 hr in a buffer solution. Over a period of 24 hr the release was about 35%. However, when the dextran capsules were challenged with a dextranase solution, simulating the arrival of the drug delivery system to the colon, the capsules quickly broke and the drug was released as a dose dump. Dextran ester prodrugs of metronidazole have been prepared and characterized for colon-specific drug release [244-246]. The kinetics of hydrolysis of metronidazole monosuccinate dextran ester conjugates in aqueous solution over the pH range 6.6–7.8 revealed that the degradation of dextran conjugates proceeds through parallel formation of metronidazole and metronidazole monosuccinate, respectively. The regeneration rates of the latter compounds followed first-order kinetics. Dextran ester prodrugs of 5-ASA were prepared and drug release rate study revealed that no drug was released in the presence of contents of small intestine of rats while the presence of diluted caecal contents accelerated drug release [247]. Dextran-nalidixic acid ester (dextran-NA) with a varied degree of substitution was synthesized as a colon-specific prodrug of nalidixic acid [248]. No drug was detected during the 6 hr of the incubation period of prodrug in buffer of pH 1.2 and 6.8, which indicated that dextran-NA might be chemically stable during the transit through the GIT. When dextran-NA was incubated with caecal contents of rats at 37oC, the extent of drug released in 24 hr was considerably increased. NA was not liberated from the incubation of dextran-NA with the homogenate of tissue and contents of the small intestine.
2.4.2 CYCLODEXTRINS
Cyclodextrins (CyDs) are cyclic oligosaccharides consisting of six to eight glucose units joined through α-1,4 glucosidic bonds (Figure 6). CyDs remain intact during their passage through stomach and small intestine. However, in the colon, they undergo fermentation from the presence of vast colonic microflora into small saccharides and thus absorbed from these regions [119,249,250]. CyDs form inclusion complexes with drug molecules because the interior of the molecule is relatively lipophilic while the exterior is hydrophilic [251,252]. It has been investigated through a study in healthy human volunteers that β CyDs are degraded to a very small extent in the small intestine but are completely digested in large intestine. Most bacterial strains that are isolated from human beings are capable of degrading CyDs. This has been proved by their ability to grow on cyclodextrins by utilizing them as the sole carbon source and by the stimulation of cyclodextrinase activity as low as 2–4 hr of exposure to CyDs. This property of the drug may be exploited for the formation of colon targeted drug delivery systems. Several CyD conjugates have been prepared and the enantio selective hydrolysis has been described [253-255]. Formulation of prodrug of CyDs with drug molecules can provide a versatile means for construction of not only colon targeted delivery systems, but also delayed release systems. Biphenylacetic acid (BPAA), an anti-inflammatory drug, was conjugated with α-, β- and γ -CyDs and in vivo release pattern was investigated in rat after oral administration. The CyD prodrug maintained the physical integrity in stomach and small intestine which is reflected from the observations that the absorption of prodrugs from upper GIT was negligible, and after 6 hr most of the prodrug approached the caecum and colon. The α- and γ -CyD amide prodrugs were hydrolyzed to the maltose conjugate in the colon while the α- and γ -CyD ester prodrugs produced BPAA in the caecum and colon. The anti-inflammatory effect of the α- and γ -CyD ester prodrugs was assessed and compared with those of BPAA alone and the drug CyD complex prepared by the kneading method in a molar ratio of 1:1. In the case of β-CyD complex, a rapid anti-inflammatory response was observed from the small intestine after a fast dissolution of the complex. In sharp contrast, the γ -CyD ester prodrug required a fairly long lag time to exhibit the drug activity, because BPAA was produced after the prodrug had reached the caecum and colon [256,257]. Hiramaya et al. [258] prepared two CyD conjugates: ester and amide conjugates. They showed that ester conjugate released the drug preferentially when incubated with the contents of caecum or colon, whereas no appreciable drug release was observed on incubation with the contents of either stomach or intestine in intestinal or liver homogenates or in rat blood. Systemic side effects of prednisolone were significantly reduced when conjugated with α-CyD [259]. The lower side effect of the conjugate was attributed to passage of the conjugate through the stomach and small intestine without significant degradation or absorption, followed by the degradation of the conjugate site-specifically in the large intestine.
2.4.3 CURDLAN
Curdlan is a neutral, essentially linear (1"3)-b-glucan which may have a few intra- or inter-chain (1"6) linkages (Figure 6). Curdlan’s unusual rheological properties among natural and synthetic polymers underlie its use as a thickening and gelling agent in foods. Apart from being tasteless, colourless and odourless, the main advantages are that in contrast to cold-set gels and heat-set gels, the heating process alone produces different forms of curdlan gel with different textural qualities, physical stabilities and water-holding capacities. Gels of variable strength are formed depending on the heating temperature, time of heat-treatment and curdlan concentration. The safety of curdlan has been assessed in animal studies and in vitro tests and it is approved in food use in Korea, Taiwan and Japan as an inert dietary fibre. It is registered in the USA as a food additive [260]. When rats were feeded with curdlan, the caecal mass increased significantly as the amount of SCFA and lactic acid in the caecal content [261]. The significant increase in the mass of the caecum was accompanied by a decrease in faecal mass. The transit time of the gastrointestinal tract was extended by curdlan supplementation [262]. Significant decrease was observed in the total hepatic cholesterol and low values were measured in the proportion with secondary bile acids. All those parameters revealed that curdlan is easily degraded and fermented by intestinal bacteria in the caecum and lowers cholesterol concentration in the liver [261]. But still bacterial degradation studies of curdlan as a polysaccharide for colon-specific drug delivery yet to be reported.
2.5 POLYSACCHARIDES FROM ALGAL SOURCE
2.5.1 ALGINATES
Alginates, natural hydrophilic polysaccharide derived from seaweed, consist of 1→4, linked D-mannuronic acid and L-glucuronic acid residues (Figure 6). Alginates are easily gelled in presence of a divalent cation as calcium ion. The gelation or crosslinking is due to the stacking of the glucuronic acid blocks of alginate chains. Calcium alginates beads can be prepared by drop-wise addition of the solution of sodium alginate into the solution of calcium chloride (Figure 6). The alginate beads have the advantage of being nontoxic, and dried alginate beads reswell in presence of dissolution media and can act as controlled release systems. Calcium alginate beads were prepared as cores and 5-ASA was spray-coated on them [263]. Different enteric as well as sustained release polymers were applied as coat on calcium alginate beads. A system was prepared by coating calcium alginate beads with Aquacoat® that is a pH-independent polymer followed by 2% w/w coating of Eudragit L-30D. Being enteric polymer, Eudragit® resisted the release of drug in acidic media and drug release was triggered at alkaline pH and controlled by thickness of Aquacoat®. When drug-loaded calcium alginate beads swell sufficiently (osmotic gradient) to exceed the strength of outer sustained released coat, the film bursts to release the drug. Such a system delivers drug to the distal intestine with minimal initial leak and provides sustained release in the colon. Kiyoung, Kun, and Yueim [264] prepared alginate beads and coated them with dextran acetate. In the absence of dextranase, minimal drug release occurred whereas it was significantly improved in the presence of dextranase.
2.6 POLYSACCHARIDES FROM FUNGAL SOURCE
2.6.1 SCLEROGLUCAN
Natural polysaccharides, as well as their derivatives, represent a group of polymers that are widely used in pharmaceutical formulations and in several cases their presence plays a fundamental role in determining the mechanism and rate of drug release from the dosage form. Among these macromolecules, scleroglucan (Sclg) also seems to be potentially useful for the formulation of modified release dosage forms and numerous studies have been devoted to this specific topic [265]. Scleroglucan (Sclg) is a branched homopolysaccharide consisting of a main chain of (1-3)-linked b-D glucopyranosyl units bearing, every third unit, a single b-D-glucopyranosyl unit linked (1-6) (Figure 6). This polysaccharide is resistant to hydrolysis and its solutions show an interesting rheological behavior: viscosity remains practically constant, even at high ionic strength, up to pH-12 and to 90oC.
Interest in this polysaccharide was first aroused in 1967 [266]. Sclg is a general term used to designate a class of glucans of similar structure produced by fungi, especially those of the genus Sclerotium. The commercial product is termed Scleroglucan, but it is also known with other names according to the company that produces the polysaccharide (e.g., Actigum, Clearogel, Polytetran, Polytran FS, Sclerogum). Because of its peculiar rheological properties and its resistance to hydrolysis, temperature and electrolytes, Sclg has various industrial applications, especially in the oil industry for thickening, drilling muds and for enhanced oil recovery and in food industry [267-269]. In pharmaceutical products, Sclg may be used as a laxative [270] in tablet coatings [271] and in general to stabilize suspensions. The use of Sclg as an antitumor, antiviral and antimicrobial compound has also been investigated [272-275]. Sclg has shown immune stimulatory effects [276] compared with other biopolymers, and its potential contribution to the treatment of many diseases should be taken into account in therapeutic regimens still Sclg as a successful agent for colonic disorders is to be investigated.
3. CONCLUSIONS AND FUTURE PERSPECTIVES
In conclusion, polysaccharides appear to be promising agents for obtaining colon-specific drug delivery systems. This article has described the different polysaccharides that have already been used in the initial approaches for colon specific drug delivery. These natural polymers are strongly appealing to use in a truly colon-specific commercially available drug delivery system. The reasons for this are that they are nontoxic, easy to work with, and will be FDA approved. Also very important is that they are selectively degraded in the colon. The challenges in the future will be to find a polysaccharide from which one might be able to obtain a non-permeable film or coating and at the same time a high degradability. These might themselves show contradicting properties. Polysaccharides with low water solubility have a better capability of retaining a drug, but at the same time degradability will be low. Because the enzymes of the colon are inducible, it may be possible to induce the necessary enzymes by giving the polysaccharide in advance prior to intake of the drug delivery system or by the sole presence of the drug delivery system. As mentioned before many bacteria adhere to particulate material and selectively induce enzymes. However, a general enzymatic induction due to the polysaccharide content of the drug delivery device is not likely to occur because the amount of polysaccharide would be very low. It is, however, necessary to obtain more knowledge of the bacterial flora and whether a more or less permanent induction of an enzyme might change the properties and ecosystem of the colonic flora in undesired directions. Finally, there is a need for obtaining standardized in vitro methods for evaluation of degradation and release as well as for an evaluation of animal models for two purposes: first, to allow comparison of results from different laboratories and second, to allow for a better correlation to the in vivo conditions in humans.
ACKNOWLEDGEMENTS
One of the Authors, Mr. Anekant Jain, is thankful to Indian Council of Medical Research (ICMR) New Delhi India for providing financial assistance to carry out project on colon targeting of drugs.
{kind=link}