J Pharm Pharmaceut Sci (www.cspscanada.org) 10(3):388-404, 2007
Methods of Endotoxin Removal from Biological Preparations: a Review
Pérola O. Magalhães1†, André M. Lopes, Priscila G. Mazzola3, Carlota Rangel-Yagui2, Thereza C. V. Penna3, Adalberto Pessoa Jr.3
1School of Health Sciences, University of Brasília 2Department of Pharmacy, School of Pharmaceutical Sciences, University of São Paulo. 3Department of Biochemical and Pharmaceutical Technology, School of Pharmaceutical Sciences, University of São Paulo
Received, December 18, 2006; Revised, April 28, 2007; Accepted, May 6 2007, Published May 18, 2007
Corresponding Author: Pérola de Oliveira Magalhães Universidade de Brasília/UnB, Faculdade de Ciências da Saúde, Departamento de Farmácia. Campus Universitário Darcy Ribeiro, Asa Norte, Brasília/DF - Brasil Phone: (55 61) 33072979 E-mail: perolamagalhaes@unb.br
ABSTRACT - PURPOSE: Endotoxins, also called lipopolysaccharides (LPS), are major contaminants found in commercially available proteins or biologically active substances, which often complicate study of the biological effects of the main ingredient. The presence of small amounts of endotoxin in recombinant protein preparations can cause side effects in host organism such as endotoxin shock, tissue injury, and even death. Due to these reactions, it is essential to remove endotoxins from drugs, injectables, and other biological and pharmaceutical products. An overview of this subject is provided by this article. METHODS: An extensive review of literature with regard to methods for removal of endotoxin from biotechnological preparations was carried out. RESULTS: A short history of endotoxin is presented first. This is followed by a review of chemical and physical properties of endotoxin and its pathophysiological effects when the body is exposed to LPS excessively or systemically. The techniques of endotoxin determination and interaction of endotoxin with proteins is also presented, taking into consideration the established techniques as well as the state of the art technology in this field. A review of techniques of endotoxin removal from biotechnological preparations is described, emphasizing how endotoxin removal can be carried out in an economical way based on a number of processes discussed in the literature (e.g., adsorption, two-phase partitioning, ultrafiltration and chromatography). Different methods are mentioned with relatively high protein recoveries; however, special attention is given to two-phase aqueous micellar systems, which are valuable tools for endotoxin removal from pharmaceutical proteins on a small scale because they provide a mild environment for biological materials. CONCLUSIONS: Efficient and cost-effective removal of endotoxins from pharmaceutical and biotechnology preparations is challenging. Despite development of novel methods, such as the two-phase aqueous micellar systems, in recent years, more research is needed in this field.
Introduction
Endotoxins are lipopolysaccharides (LPS) derived from cell membrane of Gram-negative bacteria and are responsible for its organization and stability. In pharmaceutical industries it is possible to find endotoxins during production processes or in the final product. Although endotoxins are linked within the bacterial cell wall, they are continuously liberated into the environment. The release does not happen only with cell death but also during growth and division. Since bacteria can grow in nutrient poor media, such as water, saline, and buffers, endotoxins are found almost everywhere. A single Escherichia coli contains about 2 million LPS molecules per cell. Endotoxin elicits a wide variety of pathophysiological effects. In conditions where the body is exposed to LPS excessively or systemically (as when small concentrations of LPS enter the blood stream), a systemic inflammatory reaction can occur, leading to multiple pathophysiological effects, such as endotoxin shock, tissue injury, and death (1-3). However, endotoxin does not act directly against cell or organs but through activation of the immune system, especially through monocytes and macrophages, with the release of a range of pro-inflammatory mediators, such as tumor necrosis factor (TNF), interleukin (IL)-6 and IL-1. Pyrogenic reactions and shock are induced in mammals upon intravenous injection of endotoxin at low concentrations (1 ng/mL) (4). The maximum level of endotoxin for intravenous applications of pharmaceutical and biologic product is set to 5 endotoxin units (EU) per kg of body weight per hour by all pharmacopoeias (5). The term EU describes the biological activity of an endotoxin. For example, 100 pg of the standard endotoxin EC-5 and 120 pg of endotoxin from Escherichia coli O111:B4 have activity of 1 EU (6). Meeting this threshold level has always been a challenge in biological research and pharmaceutical industry (7, 8).
In the biotechnology industry, Gram-negative bacteria are widely used to produce recombinant DNA products such as peptides and proteins. Many recombinant proteins are produced by the Gram-negative bacteria Escherichia coli. These products are always contaminated with endotoxins (6). For this reason, proteins prepared from Gram-negative bacteria must be as free as possible of endotoxin in order not to induce side effects when administered to animals or humans. However, endotoxins are very stable molecules, resisting to extreme temperatures and pH values in comparison to proteins (6, 8). Many different processes have been developed for the removal of LPS from proteins based on the unique molecular properties of the endotoxin molecules. These include LPS affinity resins, two-phase extractions, ultrafiltration, hydrophobic interaction chromatography, ion exchange chromatography, and membrane adsorbers. These procedures provide different degrees of success in the separation of LPS from proteins, which is highly dependent on the properties of the protein of interest (9).
The objective of this review is to discuss relevant aspects regarding endotoxin removal techniques from biotechnological preparations, considering its chemical and biological properties. Special attention will be given to removal by aqueous two-phase micellar systems using the surfactant Triton X-114. This review does not concentrate on the extracorporeal removal of endotoxin in vivo, which is the subject of other reviews (10-14).
HISTORY OF ENDOTOXIN
Studies about the occurrence of fever after intravenous administration of certain solutions are dated before the 19th Century. In 1894, Sanarelli showed that liquid cultures of Eberth bacillus, free from microorganisms, could produce an intoxication accompanied by fever when injected in animals, sometimes even lethal (15). By the end of the 19th Century, the designation “injection fever” was generally used to express the fever reactions observed after intravenous administration of several solutions. The administration of pharmaceuticals via intravenous route in the 20th Century increased the number of such accidents, leading several researchers to develop series of evaluating works about this subject.
In 1912, Hort and Penfold created the name “pyrogenic” to designate the “waters” which, when injected, cause “hyperthermia”. Such designation was retaken further, in 1923, by Florance Seibert, who called pyrogenic the “hyperthermizing” substances, which contained either dead bacteria – intact or disintegrated, pathogenic or not – or more often the bacterial metabolic products, such as the denaturized protein, endotoxins or exotoxins (16, 17).
The term pyrogen became popular after frequent use by Seibert, and for that reason often its creation is attributed to him (17). Seibert and co-workers continued the research started by Hort and Penfold, isolating a living Gram-negative microorganism from distillated water, which was able to produce pyrogens (15). The authors designated this microorganism as Pyrogenic bacterium, realizing that it was not a new bacterium since several varieties of microorganisms could produce pyrogens (16).
The major impulse given to increase the knowledge about pyrogens occurred between 1925 and 1945. In particular, Co-Tui, helped by Schrift, deserves special credits for showing that Gram-negative bacteria are the most dangerous producers of pyrogens. (18). It does not mean that Gram-positive bacteria cannot generate such molecules; however, they do in a lower level. In fact, Gram-positive bacteria, when destroyed by heat, produce almost no pyrogen, since in such bacteria exotoxins of proteic origin are generally formed, thus being easily denaturalized by heat. On the other hand, Gram-negative bacteria usually generate endotoxins composed mainly of lipopolysaccharides and, therefore, are more heat resistant than the first ones.
According to Westphal (1945), the pyrogens, which shall really be feared in the pharmaceutical preparations, correspond to the endotoxins of Gram-negative bacteria, and such lipopolysaccharide complexes are found in the outer layer of the bacterial cell wall (19). Essentially, pyrogens are originated in the microorganisms from the Enterobactereaceae family and are thought to be the main contaminant of an injectable solution prepared without the proper disinfecting and sterilizing processes. About two decades later, a collaborative study was developed by the US National Institutes of Health and 14 pharmaceutical industries to establish an animal system which would be adequate to evaluate the “pyrogenicity” of solutions. Such study culminated in the development of the first official pyrogen test in rabbits, which was incorporated in USP XII, in 1942. In parallel, other efforts to purify and characterize the endotoxins have taken place, and isolated pyrogenics were obtained by several researchers (17, 19)
Shear and Turner (1943) were the first researchers to use the term lipopolysaccharide to name the endotoxin extract, a term that describes the nature of the endotoxin, and that has been adopted by the scientific community (20). In 1954, Westphal and collaborators detailed the use of water-phenol systems for the production of purified lipopolysaccharides (LPS), free of proteins, from several Enterobacteriaceae. (15, 21, 22). Consequently, in recent years great progress has been made in understanding the molecular organization and mechanisms underlying the detrimental and beneficial activities of endotoxins (8, 23).
ENDOTOXIN: CHEMICAL AND PHYSICAL PROPERTIES
Endotoxins, also called lipopolysaccharides (LPS), are a major component of the outer membrane of Gram-negative bacteria (Figure 1). They are composed of a hydrophilic polysaccharide moiety, which is covalently linked to a hydrophobic lipid moiety (Lipid A) (Figure 2) (3, 6, 24). LPS from most species is composed of three distinct regions: the O-antigen region, a core oligosaccharide and Lipid A (LipA) (Figure 2).

Figure 1: Molecular model of the inner and outer membranes of E. coli K-12 according to Raetz et al., 1991 (24). Geometric form: ovals and rectangles represent sugar residues, as indicated, whereas circles represent polar head groups of various lipids. Abbreviation: PPEtn (ethanolamine pyrophosphate); LPS (lipopolysaccharide); Kdo (2-keto-3-deoxyoctonic acid).
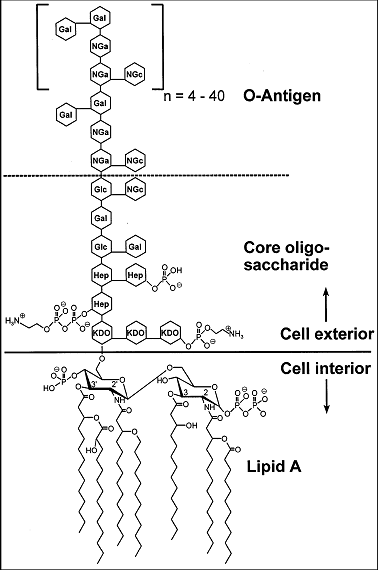
Figure 2: Chemical structure of endotoxin from E. coli O111:B4 according to Ohno and Morrison 1989 (25). (Hep) L-glycerol-D-manno-heptose; (Gal) galactose; (Glc) glucose; (KDO) 2-keto-3-deoxyoctonic acid; (NGa) N-acetyl-galactosamine; (NGc) N-acetyl-glucosamine.
The lipid A is the most conserved part of endotoxin (8, 26) and is responsible for most of the biological activities of endotoxin, i.e. its toxicity. Endotoxin is composed of b-1,6-linked D-glucosamine residues, covalently linked to 3-hidroxy-acyl substituents with 12-16 carbon atoms via amide and ester bonds. These can be further esterified with saturated fatty acids. This hydrophobic part of endotoxin adopts an ordered hexagonal arrangement, resulting in a more rigid structure compared to the rest of the molecule (8, 9). Strains lacking lipid A or endotoxin are not known. The core oligosaccharide has a conserved structure with an inner 3‑deoxy-D-manno-2-octulosonic acid (KDO) - heptose region and an outer hexose region. In E. coli species, five different core types are known, and Salmonella species share only one core structure. The core region close to lipid A and lipid A itself are partially phosphorylated (pK1=1.3, pK2 = 8.2 of phosphate groups at lipid A), thus endotoxin molecules exhibit a net negative charge in common protein solutions (8, 27). The O-antigen is generally composed of a sequence of identical oligosaccharides (with three to eight monosaccharides each), which are strain specific and determinative for the serological identity of the respective bacterium (8).
The molar mass of an endotoxin monomer varies from 10 to 20 kDa, owing to the variability of the oligosaccharide chain; even extreme masses of 2.5 (O-antigen-deficient) and 70 (very long O-antigen) kDa can be found. It is well known that endotoxins form various supra-molecular aggregates in aqueous solutions because of their amphipathic structures. These aggregates result from non-polar interactions between lipid chains as well as of bridges generated among phosphate groups by divalent cations (1). The aggregate structures have been studied by numerous techniques such as electron microscopy, X-ray diffraction, FT-IR spectroscopy, and NMR. Results from these studies have shown that, in aqueous solutions, endotoxins can self assemble in a variety of shapes, such as lamella, cubic, and hexagonal inverted arrangements, with diameters up to 0.1 mm and 1000 kDa, and high stability depending on the solution characteristics (pH, ions, surfactants, etc) (28, 29). It is proposed that proteins may also shift equilibrium by releasing endotoxin monomers from aggregates (6, 8). According to molecular dynamics, the three-dimensional structure of endotoxin, especially the long surface antigen, is much more flexible than the globular structure of proteins (8).
Endotoxins are shed in large amount upon cell death as well as during growth and division. They are highly heat-stable and are not destroyed under regular sterilizing conditions. Endotoxin can be inactivated when exposed at temperature of 250º C for more than 30 minutes or 180º C for more than 3 hours (28, 30). Acids or alkalis of at least 0.1 M strength can also be used to destroy endotoxin in laboratory scale (17).
MECHANISM OF ENDOTOXIN ACTION
Endotoxin elicits a wide variety of pathophysiological effects, such as endotoxin shock, tissue injury, and death (3). Endotoxins do not act directly against cells or organs but through activation of immune system, especially the monocytes and macrophages, thereby enhancing immune responses. These cells release mediators, such as tumour necrosis factor, several interleukins, prostaglandins, colony stimulating factor, platelet activating factor and free radicals (31, 32). The mediators have potent biological activity and are responsible for the side effects upon endotoxin exposure. These include alterations in the structure and function of organs and cells, changes in metabolic functions, increased body temperature, activation of the coagulation cascade, modification of hemodynamics and induction of shock. Many attempts have been made to prevent or treat the deleterious effects of endotoxins on immune cells, such as the use of anti-endotoxin antibodies, and endotoxin partial structures for blocking endotoxin receptor antagonists. Nevertheless, the interaction of endotoxins with immune cells is not only mediated by specific receptors. Cell priming may also occur by non-specific intercalation of endotoxin molecules into the membranes of the target cells (33).
Finally, it should be mentioned that endotoxins may also have beneficial effects. They have been used in artificial fever therapy, to destroy tumors and to improve, non-specifically, the immune defense. The uncertainty about its role for the human health was once described by Bennett (34). On the other hand, any superfluous endotoxin exposure must be strictly avoided to prevent complications. This is especially true for intravenously-administered medicines.
TECHINIQUES OF ENDOTOXIN DETERMIN -ATION
The commonly used FDA-approved techniques for endotoxin detection are the rabbit pyrogen test and Limulus Amoebocyte Lysate (LAL) assay (35, 36). The rabbit pyrogen test, developed in the 1920s, involves measuring the rise in temperature of rabbits after intravenous injection of a test solution. Due to its high cost and long turnaround time, the use of the rabbit pyrogen test has diminished, and is now only applied in combination with the LAL test to analyze biological compounds in the earlier development phase of parenteral devices. Today the most popular endotoxin detection systems are based on LAL, which is derived from the blood of horseshoe crab, Limulus polyphemus, and clots upon exposure to endotoxin. The simplest form of LAL assay is the LAL gel-clot assay. When LAL assay is combined with a dilution of the sample containing endotoxin, a gel will be formed proportionally to the endotoxin sensitivity of the given assay. The endotoxin concentration is approximated by continuing to use an assay of less sensitivity until a negative reaction (no observable clot) is obtained. This procedure can require several hours (5, 36). The concentration of 0.5 EU/mL was defined as the threshold between pyrogenic and non-pyrogenic samples (17, 36).
In addition to the gel-clot technique, manufacturers have also developed two other techniques: turbidimetric LAL technique and the chromogenic LAL technique. These newer techniques are kinetic based, which means they can provide the concentration of endotoxin by extracting the real-time responses of the LAL assay. Turbidimetric LAL assay contains enough coagulogen to form turbidity when cleaved by the clotting enzyme, but not enough to form a clot (37). The LAL turbidimetric assay, when compared to the LAL gel-clot assay, gives a more quantitative measurement of endotoxin over a range of concentrations (0.01 EU/mL to 100.0 EU/mL.). This assay is based on the turbidity increase due to protein coagulation related to endotoxin concentration in the sample. The optical densities of various test-sample dilutions are measured and correlated to endotoxin concentration helped by a standard curve obtained from samples with known amounts of endotoxin (38). A kinetic chromogenic substrate assay differs from gel-clot and turbidimetric reactions because the coagulogen is partially or completely replaced by a chromogenic substrate (39). When hydrolyzed by the pre-clotting enzyme, the chromogenic substrate releases a yellow-colored substance known as p-nitroaniline. The time required to attain the yellow substance is related to the endotoxin concentration (40). However, kinetic turbidimetric and chromogenic tests, although more accurate and faster than the gel-clot, can not be used for fluids with inherent turbidity such as blood and yellow-tinted liquids, e.g. urine, and their performance may be compromised by any precipitation from solution (37). Therefore, different methods for detection of endotoxin in different samples have been studied (37, 41).
INTRACTIONS OF ENDOTOXINS WITH PROTEINS
A number of biomolecules show interactions with endotoxins, such as lipopolysaccharide-binding protein (LBP), bactericidal/permeability-increasing protein (BPI), amyloid P component, cationic protein (42, 43), or the enzyme employed in the biological endotoxin assay (anti-LPS) factor from Limulus amebocyte lysate (LAL) (44). These proteins are directly involved in the reaction of many different species upon administration of endotoxin (45, 46). Molecular recognition can be assumed as interactions with anti-endotoxin antibodies and proteineous endotoxin receptors (e.g. CD14, CD16, CD18) (47). Other proteins interact with endotoxins even having no strong links to a biological mechanism, such as lysozyme (25) and lactoferrin (48), which are basic proteins (pI>7), electrostatic interactions can be assumed as the main driving force. Regardless of the mechanism that proves to be most significant, these interactions result in hiding endotoxin molecules, and consequently these molecules are not removed in the removal procedures. A typical example is described by Karplus et al. (49).
However, other mechanisms must exist as interactions with neutral hemoglobin (50) and even acidic proteins (pI<7) are known, taking place also at low ionic strength. It is still controversially discussed how these interactions occur. Generally, hydrophobic interactions with proteins are conceivable. However, there is no strong evidence that it drives the interaction mechanism. It is more probable that competition of protein-bound carboxylic groups and endotoxin-bound phosphoric acid groups for Ca2+ may result in dynamically stable calcium bridges between proteins and endotoxins (8).
The fact that LPS forms micellar aggregates that are considered the biologically active forms of LPS (51) could indicate that multiple proteins interact with LPS molecules. Ma et al. (2006) (52) suggested an alternative aggregation form, where the self-assembly of lipophorin particles, a protein that serves as pro-coagulant (53-55), into globular structures are the result of oligomeric interactions. This may provide cage-like coagulation products, where the lipid moiety forms a protective layer that separates the toxin from interaction with the surrounding environment.
Due to protein–endotoxin interactions, endotoxin removal from protein solutions requires techniques that are able to generate strong interactions with endotoxins, such as affinity chromatography. Alternatively, a specific dissociation of protein–endotoxin complexes may improve the availability of endotoxin molecules for removal. In view of the large variety of products, it is not possible to develop one general method for endotoxins removal from all products.
TECHNIQUES OF ENDOTOXIN REMOVAL
The question about how endotoxin removal can be carried out in an economical way has attracted the attention of many investigators and has been – although not published – the reason for process rearrangements in many cases. However, this issue has not yet been resolved satisfactorily. The discussion of relevant aspects of endotoxin removal from biological preparations and a critical review of the existing approaches are mandatory in order to develop more refined methods in the future.
In the pharmaceutical industry several alternative routes are known to generate products with low-endotoxin levels. However, their diversity indicates a dilemma in endotoxin removal. Several procedures were developed for pharmacoproteins, taking advantage of the characteristics of the production process, tailored to suit specific product requirements. Therefore, each procedure addresses the problem in a completely different way; none of them turns out to be broadly applicable. Anionic-exchange chromatography, for example, is potentially useful for the decontamination of positively-charged proteins, such as urokinase (56). However, decontamination of negatively-charged proteins would be accompanied by a substantial loss of the product due to adsorption (27, 57). For small proteins, such as myoglobin (Mr ~ 18000 Da), ultrafiltration can be useful to remove large endotoxin aggregates. With large proteins, such as immunoglobulins (Mr ~ 150000 Da) ultrafiltration would not be effective. In addition, ultrafiltration would fail if interactions between endotoxins and proteins cause endotoxin monomers to permeate with proteins pick-a-pick through the membrane.
Endotoxins can be considered to be temperature and pH stable, rendering their removal as one of the most difficult tasks in downstream processes during protein purification (58, 59). The removal of endotoxins becomes more challenging when associated with labile biomolecules, such as proteins (60). A number of approaches are typically utilized to reduce endotoxin contamination of protein preparations, including ion-exchange chromatography (61, 62), affinity adsorbents, such as immobilized L-histidine, poly-L-lysine, poly(γ-methyl L-glutamate), and polymyxin B (57, 63, 64), gel filtration chromatography, ultrafiltration, sucrose gradient centrifugation (65), and Triton X-114 phase separation (66, 67). The success of these techniques in separating LPS from proteins is strongly dependent on the properties of the target protein (9).
Two important factors influencing the success of any approach are the affinity of the endotoxin and protein antigen for the chromatography support or media used and the affinity of the endotoxin for the protein antigen. A third factor is how the affinity of the endotoxin for the protein can be modified by factors such as temperature, pH, detergents (surfactants), solvents and denaturants (4).
Usually, the procedures employed for endotoxin removal are unsatisfactory regarding selectivity, adsorption capacity and recovery of the protein. In the selective removal of endotoxin from protein-free solutions, it is easy to remove endotoxins by ultrafiltration taking advantage of the different sizes of the endotoxin and water, or by non-selective adsorption with hydrophobic adsorbent (68) or an anion-exchanger (69). For selective removal of endotoxin from protein solutions, it is necessary to know what is the form of the endotoxins in protein solutions.
Hirayama and Sakata (2002) (6) assumed that endotoxin aggregates form supermolecular assemblies with phosphate groups as the head group and exhibits a negative net charge because of its phosphate groups that originate from lipid A (6). These characteristics suggest that ionic interaction plays an important role in the binding between the cationic adsorbent and phosphate groups of the endotoxins. When hydrophobic adsorbents are used in protein solutions, it is suggested that there is also hydrophobic binding between the adsorbent and the lipophilic groups of endotoxins. These binding processes depend on the properties of proteins (net charge, hydrophobicity) and the solution conditions (pH, ionic strength).
Some commonly used techniques for removing endotoxin contaminants are ultrafiltration (70) and ion exchange chromatography (71). Ultrafiltration, although effective in removing endotoxins from water, is an inefficient method in the presence of proteins, which can be damaged by physical forces (72). Anion exchangers, which take advantage of the negative net charge of endotoxins, have been extensively used for endotoxin adsorption. However, when negatively charged proteins need to be decontaminated, they may co-adsorb onto the matrix and cause a significant loss of biological material. Also, net-positively charged proteins form complexes with endotoxins, causing the proteins to drag endotoxin along the column and consequently minimizing the endotoxin removal efficiency (57).
Alkanediols were shown to be effective agents for the separation of LPS from LPS-protein complexes during chromatography with ionic supports. Their effectiveness in reducing the protein complexation with LPS is dependent on (I) the size of the alkanediol, (II) the isomeric form of the alkanediol, (III) the length of the alkanediol wash, (IV) the concentration of alkanediol, and (V) the type of ionic support used, cationic or anionic. Alkanediol are non-flammable and as such are safer alternatives when compared to alcohols (ethanol or isopropanol) which have also been used to remove LPS from protein-LPS complexes (9). LPS removal is more efficient on cationic exchangers than on anionic exchangers.
In order to remove endotoxin from recombinant protein preparations, the protein solution may be passed through a column that contains polymyxin B immobilized on Sepharose 4B, in the hope that contaminating endotoxin binds to the gel. Similarly, histidine immobilized on Sepharose 4B also has the capability to capture endotoxin from protein solutions (63). Polymyxin B affinity chromatography is effective in reducing endotoxin in solutions (73). Polymyxin B, a peptide antibiotic, has a very high binding affinity for the lipid A moiety of most endotoxins (74). Karplus et al. (1987) (49) reported an improved method of polymyxin B affinity chromatography in which endotoxin could be absorbed effectively after dissociation of the endotoxin from the proteins by a nonionic detergent, octyl-β-D-glucopyranoside.
The methods mentioned above are reasonably effective for removal of endotoxins from protein solutions with relatively high protein recoveries. However, these affinity phases cannot be cleaned with standard depyrogenation conditions of strong sodium hydroxide in ethanol (75). Anspach and Hillbeck (1995) (57) revealed that these supports suffer from considerable efficiency decrease in the presence of proteins. Hence, they are not in general applicable for the above mentioned problem (57).
Membrane-based chromatography has been successfully employed for preparative separations predominantly for protein separations (76-83). Nevertheless, universal adoption of this technology has not taken place because membrane chromatography is limited by the binding capacity, which is small when compared to that of bead-based columns, even though the high flux advantages provided by membrane adsorbers would lead to higher productivity (78). Although bead-based chromatography is still predominant and affective for product elution operations, it has several inherent disadvantages for trace-impurity removal or polishing applications. Furthermore, the adsorptive binding capacity of bead-based columns used in this application is typically 3-4 orders of magnitude larger than required because columns are normally sized to achieve a desired flow rate rather than capacity. Since membrane-based systems have a distinct flow rate advantage and sufficient capacity for binding trace levels of impurities and contaminants, membrane adsorbers are ideally suited for this application. Work has been done recently using membrane chromatography to remove DNA, host cell protein (HCP) and endotoxin with reasonable success (8, 84-86).
Jann et al., (1975) (87) reported that slab-polyacrylamide gel electrophoresis in the presence of sodium dodecyl sulfate (SDS-PAGE) can be used for the separation of bacterial LPS. The authors showed that LPS molecular structures could be assigned to the separated LPS bands by correlating the electrophoretic banding pattern, as detected with periodic acid-Schiff stain, with the chromatography profile generated by gel permeation of chemically characterized carbohydrate moieties released from the LPS. While LPS obtained from rough (R) mutant bacteria, which contained a short oligosaccharides chain, exhibited only a fast-moving band, the LPS from wild-type smooth (S) strains, which had a core oligosaccharide substituted with various sizes of the O-specific polysaccharide chain, showed both fast and slow-migrating bands. On the other hand, LPS from the semirough (SR)-type bacteria containing a core oligosaccharide and truncated O-chain were detected as a fast-moving band migrating somewhat slower than R-type LPS bands. In spite of this great advance in the separation and analysis of intact LPS, the limited sensitivity of detection that resulted in the visualization of few broad and diffused LPS bands hindered the uncovering of further molecular intricacies of LPS (88). LPS from smooth and rough strains may be dispersed by surfactants such as sodium dodecyl sulfate (89, 90), Triton X-100 (91), and sodium deoxycholate (92-94). Upon removal of excess surfactant by dialysis, a more homogeneous population of particles with average molecular weight of about 5x105 to 1x106 Da is formed. Such observations suggest that hydrophobic interactions between subunits of LPS are important determinants of particle size (92).
Several methods have been used to separate the different subclasses of LPS from individual strains, with sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (87-95) and gel filtration (96) being perhaps the most successful. These methods are, however, hampered by the tendency of LPS to aggregate and by the difficulty in detecting and identifying each distinct subclass (97). Agarose-gel electrophoresis has been used for various purposes, such as to separate polysaccharides extracted from tissues, organs, and biological fluids of invertebrates and vertebrates (97-100). Furthermore, densiometric band analysis enables one to obtain quantitative evaluation of single polysaccharide species in mixtures (97).
Although common purification protocols may reduce the endotoxin content below the threshold level, an absolute guarantee cannot be given. It may happen that a batch of the final products is accidentally contaminated and fails the quality control. This product has to be discarded; reprocessing is not ruled out specifically but is a costly alternative.
TWO-PHASE MICELLAR SYSTEM
In recent years, the interest in the use of two-phase aqueous micellar systems for the purification or concentration of biological molecules, such as proteins and viruses has been growing (101-103). In these systems an aqueous surfactant solution, under the appropriate solution conditions, spontaneously separates into two predominantly aqueous, yet immiscible, liquid phases, one of which has a greater concentration of micelles than the other (101). The difference between the physicochemical environments in the micelle-rich phase and in the micelle-poor phase forms the basis of an effective separation and makes two-phase aqueous micellar systems a convenient and potentially useful method for the separation, purification, and concentration of biomaterials (101).
In their simplest realization, these systems exploit excluded-volume interactions between nonionic surfactant micelles and biomolecules. Specifically, in the phase-separated system one of the coexisting phases is rich in micelles while the order is poor in micelles (Figure 3) (104, 105). As a result, the stronger excluded-volume interactions between the nonionic surfactant micelles and the biomolecules in the micelle-rich phase drive the biomolecules preferentially into the micelle-poor phase based on their sizes (106).
Particularly for endotoxin removal, above the critical micelle concentration (CMC) of surfactants, endotoxins are accommodated in the micellar structure by non-polar interactions of alkyl chains of lipid A and the surfactant tail groups and are consequently separated from the water phase (micelle-poor phase). Surfactants of the Triton series show a miscibility gap in aqueous solutions. Above a critical temperature, the so-called cloud point, micelles aggregate to droplets with very low water content, by that forming a new phase. Endotoxins remain in the surfactant-rich phase. Through centrifugation or further increase in temperature the two-phases separate with the surfactant-rich phase being the bottom phase (66, 105, 107). If necessary, this process is repeated until the remaining endotoxin concentration is below the threshold limit. The cloud point of Triton X-114 is at 22°C, which is advantageous when purifying proteins.

Figure 3: Schematic illustration of a Triton X-114 micellar solution phase separation, upon temperature increase. Each of the resulting coexisting phases contains cylindrical micelles but at different micellar concentrations. Note also that, on average, the cylindrical micelles in the micelle-rich (bottom) phase are larger than those in the micelle-poor (top) phase.
Using Triton X-114, Adam et al. (1995) (108) showed a 100-fold endotoxin reduction in two steps with a final endotoxin content of 30 EU mg-1 and 50% loss in bioactivity of the exopolysaccharide. In addition, about 100-fold endotoxin reduction was shown by Cotten et al. (1994) (109), from plasmid DNA preparation with a final endotoxin content of 0.1 EU in 6 μg DNA.
A comparison of affinity adsorption and Triton X-114 two-phase extraction for the decontamination of the recombinant proteins cardiac troponin I, myoglobin and creatine kinase isoenzymes is described by Liu et al. (1997) (67). They concluded that phase separation was the most effective method, reducing the endotoxin content by 98-99% with remaining amounts of 2.5-25 EU mg-1, depending on the protein. However, Cotton et al. (1994) (109) observed slightly better removal efficiency with a polymyxin B sorbent.
Aida and Pabst (1990) (66) reported a method to reduce endotoxin in protein solutions using Triton X-114, in which the surfactant aids in dissociation of endotoxin from the protein, while also providing a convenient phase separation capability for removing the dissociated endotoxin. According to these same authors, phase separation using Triton X-114 was effective in reducing endotoxin from solutions of three different proteins (cytochrome c, albumin and catalase). The first cycle of phase separation reduced endotoxin contamination by 1000-fold. Further cycles of phase separation resulted in complete removal of endotoxin. The endotoxin was found in the detergent phase, and the upper aqueous phase contained the desired biomolecule. In addition to decontamination of endotoxin from protein preparation like recombinant products or monoclonal antibodies, phase separating using Triton X-114 should be useful for the removal of lipids from albumin or lipoproteins. Considering that a certain amount of surfactant always remains in the protein solution, which needs to be removed by additional adsorptions or gel filtration processes, this process leads to 10-20% product loss (66). It has been proposed that the detergent dissociates the endotoxin molecule from the protein and separates the dissociated molecule by phase separation using the physical characteristics of Triton X-114 (66). Liu et al. (1997) (67) demonstrated that the Triton X-114 phase separation was then further applied to other recombinant protein preparations. By performing three cycles of Triton X-114 phase separation, endotoxin levels in all recombinant proteins derived from E. coli were reduced by as much as 99% of the original amount. Furthermore, the immunoactivity, physical integrity, and the biological activity of the protein remained unchanged after the phase separation process. The phase separation can be repeated multiple times until endotoxin in the aqueous phase reaches a satisfactory level. In addition to its simplicity, this procedure is cost effective, especially in large scale.
Fiske et al. (2001) (4) examined a number of approaches to reduce the level of endotoxin, such as the use of the zwitterionic surfactants Zwittergent 3-12 (Z3-12) and Zwittergent 3-14 (Z3‑14) for the dissociation of endotoxin from the purified UspA2 protein and the subsequent separation of endotoxin from UspA2 using either ion-exchange or gel filtration chromatography. UspA2 protein is a potential vaccine candidate for preventing otitis media and other diseases caused by Moraxella catarrhalis (110). The approach that was proved successful for the dissociation of endotoxin from UspA2 was the replacement of the Triton X-100 by a zwitterionic surfactant. The inability of Triton X-100 to dissociate the endotoxin-UspA2 complex, despite success of both Z3-12 and Z3-14 may reside in the charge characteristics of the surfactants. Triton X-100 is a non-ionic surfactant containing no charged moieties while the Zwittergents contain zwitterionic head groups with both negatively and positively charged moieties. Most zwitterionic surfactants are effectively neutral; however, in some cases strong polarization exists (111). The charge characteristics of Z3‑12 and Z3-14 and the interaction of the surfactant with either the endotoxin and/or the protein may aid in the dissociation of the endotoxin from protein (in this case UspA2). Structural differences between the surfactants may also play a role in effective dissociation of endotoxin and protein. Whatever the mechanism, the use of the Zwittergent surfactant was proved to be quite suitable for the removal of LPS from UspA2 without disrupting the immunogenic properties of the protein. Prior to endotoxin reduction, the UspA2 preparations contained as much as 158 EU/Kg. However, following chromatography in the presence of Z3-12 Fiske et al. (2001) (4) achieved levels of approximately 0.0072 EU/Kg. The endotoxin removal process has been successfully implemented following GMP, to produce UspA2 subunit vaccine for clinical trials.
The levels of endotoxin appear to be much higher in recombinant proteins derived from soluble or cytoplasmatic fractions than in proteins derived from insoluble or inclusion bodies. This is consistent with the belief that lipopolysaccharides present in the cell wall are solubilized during the cell lysis procedure. Schnaitman (112) demonstrated that treatment of E. coli with the combination of Triton X-114, EDTA, and lysozyme resulted in solubilization of all lipopolysaccharide from the cell wall.
Reichelt et al. (2006) (59) tested whether the removal of endotoxin could be achieved during chromatography purification with the use of Triton X-114 in the washing steps. The application of 0.1% Triton X-114 in the washing steps was successful at reducing endotoxins during histidine and GST (resin GST sepharose) fusion protein purification, whereas washing steps lacking surfactant were ineffective in eliminating endotoxins. In contrast to purified materials employing the standard protocol which contained from 2500 to 34000 EU mg-1, purified recombinant proteins treated with Triton X-114 contained concentrations as low as 0.2 to 4 EU mg-1 (less than 1% of initial endotoxin content). Residual endotoxins in solubilized inclusion bodies can reach levels of 8 x 106 EU mL-1 despite the fact that endotoxin levels were found to be higher in recombinant proteins which are isolated from soluble fractions (113).
Endotoxins have been shown to form complexes with proteins of different isoelectric points (8) where electrostatic interactions are thought to be the main driving forces. As a result, the removal of endotoxins from basic proteins should prove to be more difficult than from acidic proteins (114). Reichelt et al. (2006) (59) studied whether the use of Triton X-114 in washing steps could eliminate endotoxins from proteins with a pI above 8.5. They found that washing with Triton X-114 coupled with affinity chromatography effectively removed endotoxins from negatively-charged proteins (SyCRP and NdhR). The minimal endotoxin concentration achieved was lower than 0.2 EU mg-1; protein recovery and yield were close to 100% (59).
Temperature-induced phase separation with Triton X-114 is a recent and powerful technique which efficiently separates hydrophobic and hydrophilic membrane proteins at room temperature, without denaturation (107, 115). This method was also successfully applied to the removal of endotoxin from proteins and enzymes, while retaining their normal functions (66). Because of an amphipathic character, LPS was also significantly removed from Klebsiella sp I-714 EPS (extracellular polysaccharides termed exopolysaccharides) after two extractions steps in 2% Triton X-114, with only a twofold decrease in bioactivity (108). According to the same authors, the Triton X-114 partitioning technique is fast, efficient, nondegradative, and allows a high level of detoxification of the Klebsiella sp. I-714 EPS. The separation of endotoxin and exopolysaccharides from Klebsiella sp. I-714 is difficult to achieve with techniques other than two-phase extraction. In addition, this method has also been successfully employed for the purification of an endotoxin-contaminated negatively-charged EPS from Pseudomonas solanacearum (108).
The detergents, even though they were also very effective at reducing the LPS levels, are relatively expensive, would add significant cost to a manufacturing process, and may affect the bioactivity of the protein of interest. Alternative chemicals are desired that could safely and cost effectively be used in place of the alcohols or detergents as washing agents for the separation of LPS from proteins during chromatographic unit operations. Ideally, these chemicals would be relatively inexpensive, chemically well defined, present minimal safety issues, and have minimal impact on the bioactivity of the protein in question when implemented into a process (9).
PERSPECTIVES
Taking into consideration the properties of two-phase aqueous micellar systems to remove biomolecules, this research group had some promising results, using Triton X-114 to remove endotoxins present in fermented culture of E. coli cells during the production of protein. According to the literature ([7], [58], [65], [107]), phase separation using Triton X-114 was effective in reducing endotoxin from solutions containing biomolecules, however the optimized conditions are still uncertain and requires further investigation.
ACKNOWLEDGMENT
The authors are grateful for the financial support from CNPq - Conselho Nacional de Desenvolvimento Científico e Tecnológico and FAPESP – Fundação de Apoio a Pesquisa do Estado de São Paulo.