| Are the current bioequivalence standards sufficient
for the acceptance of narrow therapeutic index drugs? Utilization of a computer simulated
warfarin bioequivalence model.
Manuscript received August 24th, 1998; Revised March 5th, 1999, Accepted March 23rd, 1999. Mark H. Friesen1 Scott E. Walker Abstract Purpose. The purpose of this computer simulation was to determine the likelihood of two bioequivalent (vs. reference) generic warfarin formulations (with varying bioavailability) passing current bioequivalence criteria against each other at varying bioavailability. Methods. A bioequivalence simulation program generated 100 warfarin bioequivalence (BE) studies with 24 patients/study. The reference formulation (R) was assigned a bioavailability of 90%. In these simulations the first generic (G1) had a bioavailability that was incrementally decreased from 90%. The second generic (G2) had a bioavailability that was incrementally increased from 90%. The bioequivalence testing was performed initially as G1 vs. R, then G2 vs. R, and finally G2 vs. G1. The tests were performed according to current criteria for therapeutic index drugs. Results. 5400 BE studies with a total of 129,600 subjects and 2,462,400 sampling times were simulated. When G1 vs. R was compared, fewer than 80% of studies passed when the relative AUC0-t ratios were 88% or less. When G2 vs. R were compared, fewer than 80% of studies passed when the relative AUC0-t ratios were 113% or greater. When Generic 2 and Generic 1 were compared fewer than 80% of studies passed when the relative AUC0-t ratios deviated from the reference by 7% or more. Discussion: Despite limitations this simulation indicates that two bioequivalent (vs. reference) generic warfarin products may not be bioequivalent to each other. Alternative methods of assessing bioequivalence are needed when more than one generic of narrow therapeutic index drug exists on the market. Introduction The Canadian Health Protection Branch (HPB) considers warfarin a narrow therapeutic index drug. The current HPB criteria for a narrow therapeutic range product requires that the 95% confidence interval (CI) of the Test-to-Reference ratio (T/R) of AUC0-t, and Cmax fall completely within the 80-125% boundary before the generic is considered bioequivalent (1). While this confidence interval requirement has never been formally adopted as a guideline, it is currently used by the HPB to assess the bioequivalence of narrow therapeutic index drugs and is also used by Provincial Formularies to judge interchangeability. This differs from the Food and Drug Administration (FDA) criteria, which require that the 90% CI of AUC0-t and Cmax to only fall within the 80-125% boundary (2). In 1997, Barr Laboratories (Pomona, NY) received FDA approval to market a generic formulation of warfarin in the United States. This has sparked much debate as to the implications of substituting generic versions for the currently available formulation of Coumadin (Dupont Pharma, Wilmington, Delaware, USA) (3-7). Substitution of Panwarfin for Coumadin has been reported to result in poor coagulation control (8). These two formulations have been reported to have equal bioavailability but differing rates of absorption as reflected by the time to achieve a peak plasma concentration (9). Under current criteria, even through peak concentrations have little effect on warfarin pharmacodynamics, these products would not be considered bioequivalent. Several issues determine the effect that a patient may experience as they are switched from the reference to a generic and vice versa. The first issue is tablet uniformity. Dupont Pharma (Dupont Pharma, Wilmington, Delaware, USA and Dupont Pharma, Mississauga, Ontario, Canada) has adopted stricter criteria (5) than are required by the United States Pharmacopea 23 (10) reducing variations in the daily dose during therapy. Generic products with broader content uniformity criteria could increase the variation seen in an individual from day to day (5). Another issue is whether the 20% allowable variation in average bioavailability would cause sub-therapeutic or toxic concentrations in some patients. There is a concern that even small changes in the concentration of the drug will cause large changes in the therapeutic/toxic effect of the drug (3). Even if an average variability of 20% in bioavailability would not produce a different average clinical effect, individual patients may have greater differences (3). This is because current guidelines consider only average bioequivalence, not individual bioequivalence or switchability (11,12). More than one generic product on the market creates the final concern. When two generic formulations exist in a market, both must be bioequivalent to the reference formulation. However, they may not be bioequivalent to each other. Therefore, a patient who was switched from one generic to a second may absorb a significantly different amount of the drug from the second generic formulation. This study utilizes a bioequivalence simulation program to examine this question. The purpose of this study was to determine if two generic products, known to be bioequivalent to a reference formulation, would also be bioequivalent to each other as bioavailability varied. Methods Data simulation For the simulation of bioequivalence data the spreadsheet BE2.XLM for Excel for Windows was used (13). This program used Excel macro functions to generate concentration-time data over 200 hours for each subject in a simulated two-period, two-treatment, two sequence cross-over study. Each study was simulated with 24 subjects and sampling times at 0, 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4, 8, 12, 24, 40, 60, 84, 100, 120, 160, 200 hours. Data used to generate a concentration time profile for each subject was based on pharmacokinetic parameters with random error assigned to the parameters within specified limits. The pharmacokinetic parameters were set to produce average warfarin concentration-time profiles (6,14). The parameters were set as follows: dose=10 mg, volume of distribution (Vd)=10 L, fraction unbound=1%, Intrinsic hepatic clearance (Clint) =120 L/h, hepatic blood flow=100 L/h, ka=1.5/hour, renal clearance=0. Both inter-individual and intra-individual variability was introduced to the Clint and Vd. Average intra-individual variability for Clint set at 10% with inter-individual variability set at 20%. Average intra-individual variability for Vd was set at 15% with inter-individual variability set at 25%. Assay variability was 10%. Bioequivalence calculation In simulating a single study, 19 sample concentration-time profiles were generated for 24 subjects for each formulation by Monte Carlo simulation. From each concentration-time profile standard bioequivalence parameters were calculated, including the AUC0-t, AUC0-¥ , Cmax, Tmax, and t1/2. Cmax was the highest observed concentration and Tmax was the time at which this highest concentration occurred. AUC0-t was calculated using the trapezoid rule. AUCt-¥ was calculated as the concentration at the 200 hour post-dose time point divided by the elimination rate constant (k). t1/2 was calculated from 0.693/k and k was calculated by least squares linear regression of the log-transformed concentration in the terminal elimination phase. For each study, the mean geometric Test-to-Reference ratio for AUC0-t, AUC0-¥ , Cmax, and the associated 95% confidence intervals were calculated using a 2-factor (subject and treatment) analysis of variance following log transformation of the data. The inter- and intra-individual variability observed in each study was calculated from the two factor analysis of variance of log transformed Cmax, AUC0-t and AUCt-¥ . One hundred studies, each with 24 subjects, were simulated following each incremental adjustment in bioavailability. The proportion of studies with a 95% CI that did not extend beyond the 80-125% limit was determined for each adjustment in bioavailability. Study Conditions In each simulation of 100 studies, the percentage of the dose absorbed (F) was adjusted to allow for different degrees of relative bioavailability. In all cases the bioavailability of the reference was maintained at 90%. Three sets of study data were generated. One set was designated Generic1 vs. Reference (G1 vs. R) in which the bioavailability of the reference formulation (R) was set at 90% and the mean bioavailability of the generic formulation (G1) was decreased from 90% at 1 % increments until all the studies failed. The second set was designated Generic2 vs. Reference (G2 vs. R). In this set the bioavailability of the reference formulation (R) was again set at 90% and the mean bioavailability of the generic formulation (G2) bioavailability was increased from 90% at 1 % increments until all 100 studies failed. The third set was designated Generic2 vs. Generic1 (G2 vs. G1). The purpose of this was to create a range of relative bioavailability that would result in a bioequivalence failure rate running from 0% to 100%. Results 5400 bioequivalence studies, each consisting of 24 subjects in a 2-way cross-over study, were simulated. In total these simulations comprised 129,600 subjects and 2,462,400 concentrations. The simulated data for the reference product produced concentration-time profiles with characteristics similar to literature values for warfarin (6,14) (Table 1). Table 1. Mean pharmacokinetic parameters of warfarin
* with bioavailability set at 90%. ** average of 5400 trials calculated from log-transformed 2-way ANOVA The simulated results generated means which were close to expected. The relative bioavailability deviated by less than 0.5% from expected, intra-subject or residual variability CV(%) averaged 12.1% and inter-subject CV(%) averaged 22.5% for AUC0-t. These intra and inter-subject variability's observed in the simulated studies are also similar to literature values. Generic 1 versus reference was observed to have more than 80% of the bioequivalence studies pass when the relative bioavailability was 88%. This study-passing rate dropped to less than 10% when the relative (G1 vs. R) bioavailability was reduced to 82% (Table 2). Generic 2 versus reference reached less than 80% and less than 10% of studies were observed to pass at relative bioavailabilities of 112% and 122%, respectively (+12% and +22%) (Table 3). Less than 80% and less than 10% of studies were observed to be bioequivalent when the relative bioavailability of generic 2 versus generic 1 reached 114 and 122% (relative to each other) (+14 and +22%), respectively (Table 4). Table 2. Summary Results for 100 Simulated Generic 1 vs. Reference Studies.
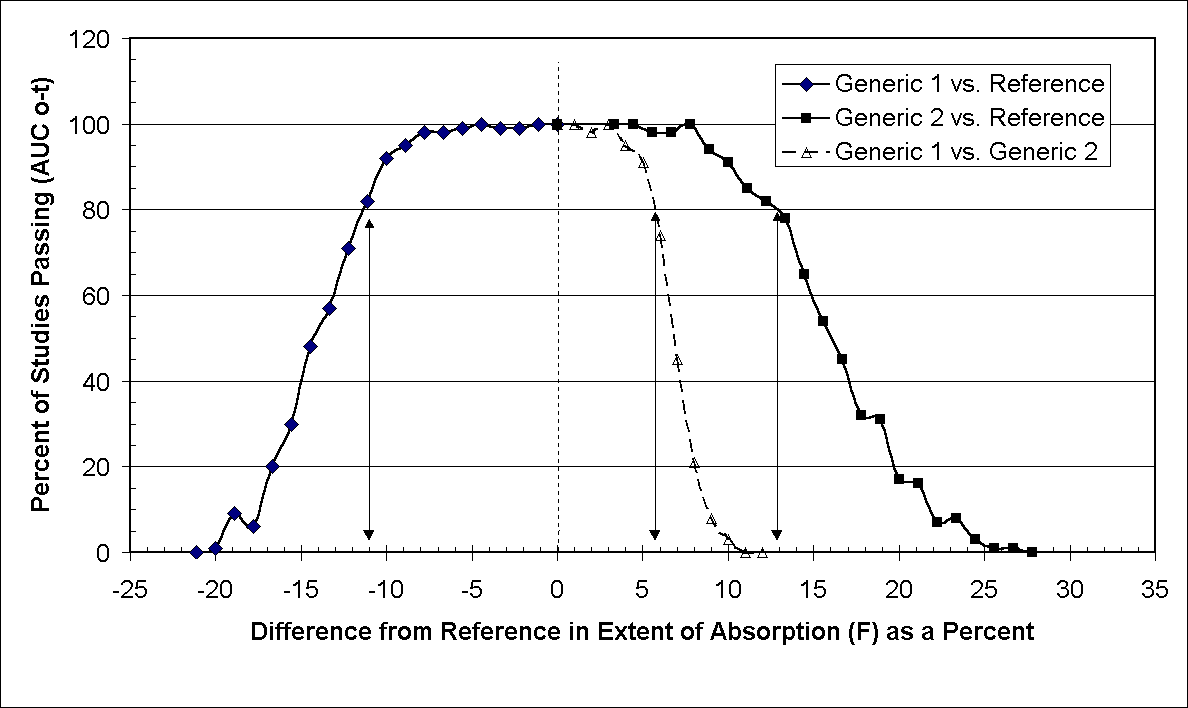
However, because G1 and G2 had their relative bioavailability simultaneously and incrementally changed in opposite directions, less than 80% of G1 vs. G2 studies would be judged bioequivalent to each other when each generic differed from the reference (R) by only 6% or more. As the difference in relative bioavailability between G1 and G2 increased to 22%, fewer than 10% of G1 vs. G2 studies were passing, yet the difference from the reference (R) was only 10 (Table 4). Under the conditions of this study, when a generic differs from the reference by only 10% and the residual variability is ~12 to 13%, approximately 90% of studies will pass (Tables 2 and 3). However, if the two generics formulations differ from the reference to an opposing degree (+10% and –10%), less than 5% of the G1 vs. G2 studies would be declared bioequivalent. These results use AUC0-t as the pharmacokinetic parameter to determine bioequivalence (Figure 1). Similar results are obtained with both AUC0-¥ , and Cmax.
Table 3. Summary Results for 100 Simulated Generic 2 vs. Reference Studies.
Figure 1. The percentage of studies that were observed to have their 95% CI fall completely between 80 and 125% as a function of the T/R ratio (%) in extent of absorption (F), expressed as the difference from reference, when reference is anchored at 1 or 100% [if FGeneric = 0.85 and FReference = 0.90, the ratio = 0.944 or 94.4% and the difference = -5.6%]. Perpendicular lines with arrows show points at which 80% of the studies have 95% CI fall completely between 80 and 125%. The open triangles identify two generic products which differ from the reference by equal but opposite bioavailabilities. When Generic 1 is -6% from reference and Generic 2 is +6% from the reference, while both would be bioequivalent to the reference (~98% of studies between Generic 1 and Reference or Generic 2 and reference would pass) only 74% of studies evaluating Generic 1 vs. Generic 2 would be found to be bioequivalent. As this difference in bioavailability increases, the failure rate also increases. When Generic 1 is -11% from the reference, and Generic 2 is +11% from the reference, more than 80% of the studies would find either generic bioequivalent to the reference. However, at this point, 0% of studies would find the generic products equivalent to each other. Discussion Current criteria for bioequivalence can result in a new generic formulation being declared bioequivalent and interchangeable with a Canadian reference product. Subsequent market entries may also be declared bioequivalent and interchangeable with the same Canadian reference product (1). However, while bioequivalence of the first generic relative to the subsequent generic market entries is never tested and never declared, in practice, interchangeability between all generic products and the reference occurs. In this situation the possibility exist for two generic products, which are known to be bioequivalent to a reference formulation, to be bio-in-equivalent to each other. In this study we tested the tolerance of the current Canadian narrow therapeutic index criteria to the bioequivalence between generic products.The results of this study suggest that two generic warfarin formulations, both of which met the criteria for bioequivalence against a reference formulation, would not necessarily meet bioequivalence criteria if compared to each other. It is clearly seen that when a difference between 6 and 11% in bioavailability exists between the generic and the reference product, a generic warfarin formulation would have ³ 80% likelihood of passing. However if the same generic product was compared to another generic, with similar but opposite difference in bioavailability, the likelihood of the two generic formulations being bioequivalent to each other would be between 74 and 3%. It is in this range of 6-11%, that the generic would likely to be bioequivalent to a reference, but unlikely to be bioequivalent to an equally but oppositely divergent generic. When the bioavailability differs by less than 6%, the generic formulations should be bioequivalent to both the reference formulation and the other generic formulation. When the difference in bioavailability is greater than 13% the generic would not likely be shown to be bioequivalent to the reference or an equally and oppositely divergent second generic. Therefore, within a certain range of relative bioavailability there is danger that a warfarin product that passes current standards of bioequivalence, relative to a reference formulation, will not be bioequivalent to other generic warfarin formulations. Table 4. Table 3 Summary Results for 100 Simulated Generic 1 vs. Generic 2 Studies
Deviation of about 6% (T/R) is realistic and represents a moderately reasonable copy of a reference product. However, where one generic is -6% (T/R) and a second is +6% (T/R), interchanging generic 1 for generic 2 may cause changes in effect which are greater than any change in effect which could occur as a result of interchanging either generic with the reference. Larger changes in concentration following substitution of one generic for another generic create a situation with greater risk to the patient. This would seem to be intuitively true for any two oppositely divergent generics given the current bioequivalence criteria, but the consequences are particularly important for narrow therapeutic index drugs. There are a number of possible ways in which this problem could be addressed. Firstly, one might argue that the current 95% confidence limits for the relative generic/reference ratio may allow the relative generic/generic ratio to remain within a 90% confidence interval. In fact this was not true in this experiment. With studies of 24-subjects and the degree of intra-subject variability (CV% ~12%) built into this study, 90% confidence intervals are approximately ± 1.2 to ± 1.5% narrower. Therefore, generics which deviate between 8 and 11% from the reference could be found to be bioequivalent to the reference (at a 95%CI) but would be bio-in-equivalent to an equally but oppositely divergent generic, even using a 90%CI. Alternatively, each new generic drug could be required to be tested for bioequivalence against all current versions on the market. However, this is an expensive alternative, especially for a reference product that has multiple generics, and could drive up the cost of generics, defeating the purpose of their existence. Thirdly, individual bioequivalence could be incorporated into the testing criteria, which may more accurately reflect "switchabilty" between products. However, while individual bioequivalence is used to measure switchability, since studies evaluating individual bioequivalence still only measure the switchability between a single generic and the reference, the problem of switchability could still exist between two generics. Therefore, to protect patients and ensure that all products fall within 95% CI criteria, an additional criterion seems necessary. From our study it would seem that limiting the geometric mean ratio (T/R) to a deviation of less than 5% would completely eliminate the problem. However, this observation is based on both a sample size of 24 and the residual variability (12%) used in the simulations. Increasing sample size and or reducing variability will reduce the failure rate for a given difference in bioavailability. This will result in a larger cut-off in the T/R ratio. The opposite effect is observed for increased variability and or smaller sample sizes. It would require additional evaluation to determine the precise effects of each parameter on the proposed T/R ratio cut-off. Nevertheless, a simple evaluation indicates that increasing sample size from 24 to 32 changes the lower limit of a 95% confidence interval by 2% and the upper limit by 3.5%. Reducing CV from 12% to 8% changes the lower limit of a 95% confidence interval by 3% and the upper limit by 4.5%. The effect of each parameter, considered separately, would cause our cut-off (the point at which generic 1 vs. generic 2 begin to fail while each generic vs. reference still passes) to shift from ~6% to ~8% and the upper range of ~13% to shift to 15%. On this basis, for a drug such as warfarin, a 5% limit still appears reasonable, achievable and should prevent clinically important differences from occurring in patients. Furthermore, the generic version of warfarin marketed by Barr Laboratories is reported to have a mean T/R ratio which is within 2% of the reference for all strengths and a 90% CI for AUC which does not deviate by more than 5.3% from the brand product. Scaling is an alternative solution to the same problem and has been recently proposed by Midha et al (15). While scaling was initially considered highly variable drugs, it is now proposed that confidence intervals be scaled for all drugs based on the observed intra-subject or residual variability. This is based on the observation that drugs with low intra-subject variability have far more license to deviate from the reference than do more variable drugs (15). Scaling to the degree indicated by Midha et al (15) would result in a 90% confidence interval having an upper bound of 110% compared to the commonly used bound of 125% for a drug such as warfarin. In this study, with an intra-subject CV averaging between 12 and 13%, when the 95% T/R confidence interval fell completely between 90 and 110%, the geometric mean ratio was deviated by 3% or less. If a 90% confidence limit is used, the geometric mean ratio between test and reference formulations would be allowed to deviate by approximately 4.5%. While the proposal of Midha et al (15) has wider application, there would appear to be similar agreement in what is regarded as an allowable deviation in T/R geometric mean ratios. There are a number of limitations to the model used to predict bioequivalence in this study. The model was simplistic in that the only variability in the CLint and the Vd was allowed. While overall variability generated in these simulations was comparable to the variability (inter and intra) observed in warfarin studies, there was no variability (intra or inter) built into the rate and extent of absorption of the product. The same is true in regard to the dosage variability. This model also only considers the situation where the intra-individual variability is 12-13% and the studies have a sample size of 24 subjects. While neither parameter affects the confidence interval, it does affect our observation that a 5% cut-off would eliminate the situation where generics might not be bioequivalent to each other but would be bioequivalent to the reference. Nevertheless, despite limitations in the model, we still believe that a 5% limit is still a reasonable, achievable and a clinically important endpoint. Conclusions It should be recognized that the data presented in this paper are based on simulation. However, despite limitations, the result does indicate that current standards would allow two generic narrow therapeutic index formulations of warfarin to be bioequivalent to brand name warfarin formulation while the two generics may not be bioequivalent to each other. This could be clinically important given the narrow therapeutic index of this drug. We therefore propose that a 5% limit in the T/R mean ratio, be added to the criteria for evaluation of narrow therapeutic index drugs. References
Corresponding author: Scott Walker, MScPhm, Coordinator, Quality Control and Research, Department of Pharmacy, Sunnybrook Health Science Centre, 2075 Bayview Avenue, North York. Ontario, Canada M4N 3M5. Email: scott.walker@sunnybrook.on.ca Keywords: Bioequivalence, Warfarin, Monte Carlo simulation, Narrow therapeutic range Published by the Canadian Society for Pharmaceutical Sciences. Copyright © 1999 by the Canadian Society for Pharmaceutical Sciences. |