J Pharm Pharmaceut Sci (www.ualberta.ca/~csps) 2(3):89-91, 1999
|
||||||
Human In Vivo Competitive Inhibition of P450 Substrates: Increased Plasma Concentrations as a Function of Hepatic Extraction Ratio and Percent Inhibition Received manuscript July 2th, 1999; Revised September 27th, 1999; Accepted November 1st, 1999. Harold Boxenbaum1 PDF Version for printing Abstract The purpose of this note is to posit and discuss the concept of “competitive inhibition potential” (CIP), which is an in vivo index of the ability of a competitive inhibitor to elevate plasma concentrations of drug substrates, when the competitive inhibitor is administered at its usual and customary dose. Consider an orally administered drug completely eliminated by the liver via a single enzyme, not undergoing efflux or gut wall metabolism, exhibiting linear pharmacokinetics, where fraction available to the liver is unaltered in the presence of a competitive inhibitor and hepatic blood flow is constant (assume 25 mL/min/kg throughout). Assuming a single-dose or constant rate of dosing, either the area under the blood/plasma concentration-time curve from zero to infinity (AUC) or the average steady-state concentration (Cav) may be expressed as follows (1): where ER is hepatic extraction ratio. The numerator reflects the contribution of first-pass hepatic metabolism, whereas the denominator reflects systemic clearance. With IV dosing (no hepatic first-pass effect), the numerator is simply set to unity. In the in vivo presence of a competitive inhibitor, percent inhibition of metabolism may be expressed as follows (1):
where Ki is the competitive inhibitor constant, and the ER subscript indicates whether ER occurs in absence or presence of competitive inhibitor. Inasmuch as serial in vivo competitive inhibitor concentrations may be sufficiently represented as a constant (e.g., Cav), the ratio of AUC or Cav in the presence of a competitive inhibitor to that in the absence of a competitive inhibitor (R) is given by (1):
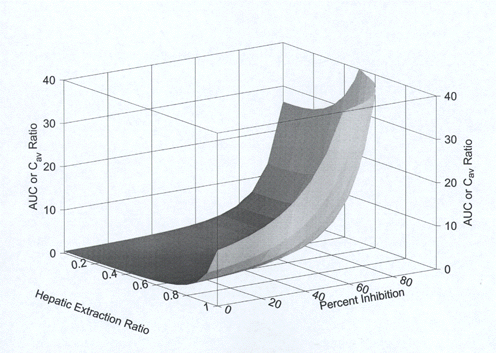
Through algebraic combination of Equations 1-3, a three dimensional plot of ER, %I and R was constructed and is illustrated in Figure 1. Visual inspection clearly indicates that drug exposure enhancement (measured by AUC or Cav) is highly dependent on both ER in the absence of inhibitor, as well as the extent of inhibition (dependent on inhibitor plasma concentration and Ki). Of particular interest is the observation that high clearance drugs (i.e., those with high ERs) are at greater risk of significant increases in exposure in the presence of an inhibitor. This helps explain why intact terfenadine (predominantly a P450 3A4 substrate) exposure increases approximately 37 fold during co-administration with high-dose ketoconazole (200 mg po Q 12 h). Its ER is reduced from ~95% (alone) to ~35% (with high-dose ketoconazole) (1). There are at least a few ways of ascertaining in vivo Ki values (1). Utilizing literature data (1-3), the Ki plasma values for ketoconazole and clarithromycin inhibition of CYP 3A4 (triazolam substrate, predominantly CYP 3A4 eliminated) in healthy human subjects have been determined as ~1.24 mg/mL and ~0.52 mg/mL, respectively. (In theory, Ki values are substrate independent in purified enzyme preparations, although this will require validation in vivo).
At ketoconazole oral doses of 200 mg once daily, Cav is ~1.0 mg/mL in healthy human subjects (4). At clarithromycin doses of 250 mg po Q 12 hours, Cav is ~0.57 mg/mL in healthy human subjects (5). Using these doses (which may vary, depending on therapeutic considerations), one may calculate a unitless “competitive inhibition potential” (CIP), as follows:
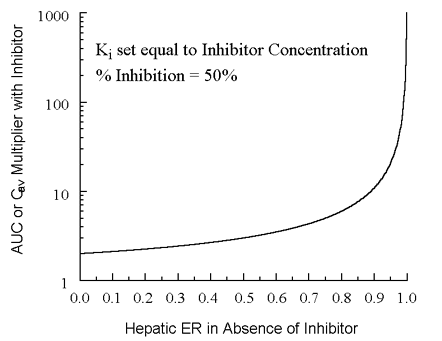
CIP values for ketoconazole and clarithromycin are ~0.8 and ~1.1, respectively. Consequently, at therapeutic doses assumed above, each will produce similar increases in CYP 3A4 substrate exposures. As noted previously (1), plots of R vs. ER (absence of inhibitor) are useful in assessing substrate exposure consequences of competitive inhibitor co-administration. Figure 2 illustrates this plot, assuming Ki = inhibitor plasma concentration, somewhat mimicking both ketoconazole and clarithromycin in the examples discussed above.
Note high ER drugs are particularly susceptible to large changes in exposure (R). CYP 3A4 substrates with high ERs exhibiting large increases in R probably include cisapride, terfenadine, midazolam, astemizole, triazolam, certain HMG Co-A reductase inhibitors, etc. Triazolam has an ER of ~0.3 (1). With 50% inhibition (Ki = inhibitor concentration), R ~ 2.4. When percent inhibition equals 73.7%, (I)/(Ki) = 2.8, ER(with inhibitor) = 0.0789, and R = 5.0. Taking clarithromycin Ki as 0.52 mg/mL (see above) with a clarithromycin average steady-state plasma concentration of 1.456 mg/mL (observed clinically with ~500 mg po every 12 hours), %I = 73.7% and R = 5. This triazolam R value was observed (2) when clarithromycin was employed as an in vivo competitive inhibitor of oral triazolam. A major objective for in vitro screening of potential competitive inhibitors of CYP 3A4 will be extrapolation of in vitro Ki data employing human microsomes, hepatocytes, etc. Although a thorough discussion of this is beyond the scope of this present work, one would probably have to consider unbound inhibitor concentrations both in vitro and in vivo. A final note on drug development of oral new chemical entities metabolized predominantly by CYP P450 enzymes: Low clearance drugs are not only preferable to increase the probability for once-daily dosing, but they will also have lower R values and will therefore be safer with competitive metabolic inhibition. Cyclosporin (ER ~0.213 in normal subjects) is a good example of a low clearance CYP 3A4 substrate. When competitive inhibitor plasma concentration equals in vivo Ki, %I is 50%, and R = 2.27 (this R is frequently observed clinically). For terfenadine with an ER of 95%, 50% inhibition would increase exposure (R value) 21-fold. Since terfenadine is strongly arrhythmogenic, this helps explain observed QT interval prolongation, torsades de points, etc. when administered with competitive inhibitors such as erythromycin and ketoconazole. References
Corresponding Author: Harold Boxenbaum, Arishel Inc., 14621 Settlers Landing Way, North Potomac, Maryland, USA, email:shelbyb@ix.netcom.com Published by the Canadian Society for Pharmaceutical Sciences. Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences. |
||||||
|