J Pharm Pharmaceut Sci (www.ualberta.ca/~csps) 3(1):137-155, 2000
Detection and Prevention of NSAID-Induced Enteropathy
Manuscript received December 10, 1999, Revised April 27th, 2000; Accepted April 28th, 2000.
Neal
M. Davies1,
Joseph Y. Saleh
Faculty of
Pharmacy, University of
Sydney, Sydney, New South Wales, Australia
Respiratory Research Group, Faculty of Medicine, University of Calgary, Calgary, Alberta, Canada
PDF version for printing
Abstract
Non-steroidal
anti-inflammatory drugs (NSAIDs) may cause damage distal to the
duodenum. We reviewed the prevalence, clinical spectrum, assessment,
pathogenesis, and treatment of adverse effects of NSAIDs on the small
intestine. NSAIDs can cause
small intestinal perforation, ulcers, and strictures requiring surgery.
NSAIDs produce inflammation of the small intestine in 40 to 70% in
long-term users, and the associated complications of blood loss and
protein loss are difficult management problems. The pathogenesis of
NSAID enteropathy is a multi-stage process involving specific
biochemical and subcellular organelle damage followed by inflammatory
tissue reaction. Various suggested treatments of NSAID-induced
enteropathy (e.g., sulphasalazine, misoprostol, and metronidazole) have
yet to undergo rigorous clinical trials. Cyclo-oxygenase-2 inhibitors
appear to be safer to the small intestine than traditional NSAIDs.
Pre-clinical and clinical data suggests meloxicam, celecoxib, nimesulide
and rofecoxib may have less small intestine toxicity than traditional
non-selective NSAIDs.
Introduction
The anti-inflammatory, analgesic, and anti-pyretic properties of NSAIDs are particularly useful in treating rheumatic and other musculoskeletal disorders. During the last fifty years a plethora of NSAIDs have been introduced on the market indicative of the commercial potential for such compounds and attesting to their utility in the treatment of pain and inflammation of varying origin from the head to the big toe. Overall, there is probably little difference in the analgesic and anti-inflammatory efficacy between the different NSAIDs when evaluated in large patient populations. Hundreds of comparative clinical trials of various NSAIDs in rheumatic disorders have confirmed this observation.[1] The impression of most clinicians prescribing NSAIDs is that these drugs provide equivalent efficacy once a sufficient dosage of any individual NSAID is used. Despite the epidemiological findings of generally equivalent therapeutic responses amongst large cohorts of patients, it is also clinically apparent that marked variability in the response of individual patients to different NSAIDs exists. Individual patients will note significant differences in efficacy between different NSAIDs. Approximately 50% of patients respond to the first NSAID tried, and 25% of initial non-responders to a second NSAID, and 10% of repeat non-responders to a third NSAID.[2]
The clinical utility of an NSAID is determined by the compromise between its therapeutic efficacy and toxicity. If an NSAID is effective, but a patient cannot tolerate its side-effects then the NSAID is of no use to the patient. Safety is often a primary consideration in the choice of a particular NSAID. Given the apparent equivalent efficacy of NSAIDs it is not surprising that the promoted “safety” of an NSAID is a main determinant of its sales success. NSAID toxicity can be reduced by avoiding their use in high-risk patients, however, this is not always practical given that many such patients may require NSAIDs for daily functioning.
A major limitation of NSAIDs' clinical utility is their gastroduodenal epithelial toxicity. NSAID toxicity is, however, not site-specific to the gastroduodenum. NSAIDs can induce toxicity in the more distal intestine.[3] There are a number of literature review articles summarizing the toxicology of NSAIDs in the gastroduodenum. This article will focus on the prevalence, clinical spectrum, assessment, pathogenesis, and treatment of adverse effects of NSAIDs on the small intestine. This article will also discuss in detail the distinct small intestine toxicology of preferential and selective COX-2 inhibitors.
Clinical
Spectrum of NSAID Side-Effects in the Small Bowel
The gastric side-effects of NSAIDs were recognized more than a century ago.[4] The first reports of NSAID toxicity in the small bowel were from laboratory animal studies demonstrating indomethacin-induced intestinal bleeding, inflammation, and ulceration.[5] In humans there is indirect evidence of small intestine NSAID toxicity. Amongst patients with bleeding upper GI lesions, ulcer healing is not always accompanied by a correction of anemia.[6] Furthermore, there is often a lack of correlation between abdominal symptoms and gastroscopic evidence of damage.[7] Such clinical observations suggest the possibility of more distal sites of damage. Case reports have been accumulating associating NSAIDs with intestinal lesions.[3,7-45]
The already identified small intestinal side-effects of NSAID use include increased intestinal permeability, diaphragm like-strictures, ulceration, perforation, hemorrhage, and death.[3] Intestinal blood loss may significantly contribute to anemia in rheumatoid arthritis patients taking NSAIDs. Indeed, it has been postulated that NSAID-induced distal intestinal and not gastroduodenal bleeding may be the major source of hemorrhage from the GI tract.[46] Vitamin B12 and bile acid absorption may also be impaired thereby further aggravating hemorrhage-induced anemia.[47-48] Studies have shown that chronic NSAID users develop small intestinal inflammation associated with both blood and protein loss that may persist up to 16 months following discontinuation of the NSAID.[24,49-50] Further studies showed that long-term NSAID treatment was associated with enhanced migration of 111indium-labelled leucocytes to the ileum, increased faecal 111indium secretion, and increased faecal calprotectin shedding.[51-52] Each of these studies adds growing evidence that NSAIDs produce clinically significant inflammation with secondary hemorrhage and protein loss.
‘Diaphragm’-like strictures associated with chronic NSAID use have been reported by several investigators.[10,21,29,34,53-54]. NSAIDs also induce narrow-based ileal stenoses.[30,55] The histopathological description of these stenoses is very characteristic, if not pathognomic, for NSAID-induced damage, and has been coined 'diaphragm disease.'[30] These 'diaphragms' are numerous, thin (2 to 4 mm), concentric, septate-like mucosal projections that narrow the intestinal lumen. They are mainly located in the mid-small intestine, and are histologically characterized by prominent submucosal fibrosis without evidence of vascular involvement.[30]
Pre-term infants are at increased risk of necrotizing enterocolitis and intestinal perforation after receiving indomethacin for patent ductus arteriosus closure.[12,16,42]
The potential prevalence and morbidity of NSAID enteropathy may rival or exceed that of the longer recognized, better described, and clinically more dramatic NSAID gastropathy. Ongoing and further investigation of the small intestinal toxicity of NSAIDs is required. Available laboratory methods to investigate NSAID entropathy will be detailed.
Techniques to Assess NSAIDs and the Small Intestine
Methods of Assessing
Gastrointestinal Permeability
The diagnosis of NSAID enteropathy is presently difficult and clinically impractical. To replace cumbersome endoscopic or surgical methods of surveying the small intestine substantial efforts have been made to develop non-invasive methods of detecting intestinal abnormalities. The intercellular junctions of intestinal epithelial cells appear to be particularly susceptible to NSAIDs. These junctions may be the amongst the first epithelial cell organelles to suffer discernable damage following disruption of intracellular energy production.[56] The resultant loss of intercellular integrity allows the permeation of passively transported, water soluble macromolecules across the gastrointestinal epithelium.
Tests of GI permeability are designed to assess the functional integrity of the intestinal epithelium. This can be done non-invasively by measuring the urinary recovery of orally administered probes. Such urine recovery assays have been found to be extremely useful in measuring gastroduodenal and intestinal NSAID damage.[56,57] In vivo intestinal permeability has been assessed by a number of analytical techniques. The three most commonly employed orally-ingested probes are carbohydrates (e.g., lactulose, cellobiose, and mannitol), ethylene glycol polymers (e.g., polyethylene glycol, PEG), and non-degradable radionuclides (e.g., 51Cr-EDTA).[58,59]
In addition to their non-invasive nature, a distinct advantage of permeability tests is that they reflect the global functional integrity of the small intestinal epithelium. Morphological analyses may suffer from sampling error- particularly if intestinal abnormalities are heterogeneously distributed.
Current interest in measurement of intestinal permeability originated from the initial work undertaken with polyethyleneglycol (PEG). Subsequently, a number of carbohydrates have been used in studies of permeability including the hexoses (i.e. L-rhamnose), sugar alcohols (i.e. D-mannitol), and the disaccharides (e.g. lactulose, cellobiose). Most currently utilized carbohydrate permeability studies employ differential sugar absorption tests, in which hypertonic solutions of two sugars (usually a mono- and a di-saccharide) are given concomitantly after an overnight fast and timed urinary recovery of each sugar subsequently determined.[59] The recovered monosaccharide reflects permeation through aqueous pores, and the recovered disaccharide reflects permeation through the intercellular pathway. The ratio of the two recovered sugars gives an index of relative function of the permeation pathways.[60]
51Cr-EDTA was initially used as a marker of glomerular filtration, and subsequently in a screening test for coeliac disease.[61,62] The permeation of 51Cr-EDTA has been shown to be relatively specific to the small intestine, and a comparison of oral to intraduodenal 51Cr-EDTA instillation showed no significant differences in the extent of urinary excretion.[63-64] More recent studies have suggested that there may also be a small degree of colonic permeation.[65-66] Bjarnason, et al. [50] have suggested that this colonic permeation is small as 51Cr-EDTA is incorporated into feces and therefore unavailable for colonic permeation. The 51Cr-EDTA test has been shown to be a reproducible, safe, simple, and accurate permeability test.[61-62]
NSAID-induced increases in intestinal permeability have been extensively studied in humans.[51, 60, 64, 79, 93,97,99] These permeability changes have been detected by the oral administration of probes such as 51Cr-EDTA, lactulose, cellobiose and polyethylene glycol (PEG)[108-111] with 51Cr-EDTA being the most frequently employed probe in NSAID-induced intestinal permeability studies.[108-111]
The available permeability data use indomethacin or naproxen as prototypical NSAIDs.[51, 60, 64, 79, 93,97,99,112] In general, these studies show a dose-dependent increase from baseline of 51Cr-EDTA excretion with NSAID administration.[64] After two doses of either acetylsalicylic acid (1.2 g twice), ibuprofen (400 mg twice) or indomethacin (75 and 50 mg) 51Cr-EDTA measured permeability increased significantly compared to drug-free controls in normal humans.[60] Increased intestinal permeability was correlated to NSAID cyclo-oxygenase inhibition. Intestinal permeability also increased equally after oral and rectal administration of indomethacin, suggesting increased permeability results from the systemic effects of this NSAID.[51]. The 51Cr-EDTA test was able to detect an increase in intestinal permeability resulting from the administration of two different doses of naproxen. There was a statistically significant difference between the median 51Cr-EDTA excretion following 750 mg naproxen (19%) and 1000 mg naproxen (68%).[64]
Enteroscopy
Gastroduodenoscopy
has been the gold standard assay for NSAID-induced epithelial damage as it
had been generally believed that GI side-effects of NSAIDs were usually
confined to the upper GI tract. Attempts to examine the more distal small
bowel endoscopically have been limited by the use of awkward instruments
and techniques. Morris et al. [47] described the use of a balloon-driven
enteroscope to obtain extended views of the small bowel. The possibility
of small bowel enteroscopy to detect small intestine NSAID-induced
strictures and lesions was subsequently been reported by several other
groups.[67-69]
Enteroscopy for diagnosis of small intestinal damage induced by NSAIDs is unsuitable as a screening test as it is time-consuming and expensive. Detection of NSAID-induced intestinal abnormalities is also associated with other difficulties such as the imprecise anatomic localization, subjective grading of lesion severity, and confusion with other intestinal diseases. Complete visualization of the small intestinal mucosa during enteroscope withdrawal may be inadequate due to the rapid passage of the enteroscope tip around intestinal loops. The passage of an enteroscope to the ileocecal valve junction may require up to 6 hours in some patients.[68] Small intestinal lesions associated with NSAIDs can only be described using only non-specific terms such as edema, erythema, mucosal hemorrhage, erosions, or ulcers. Enteroscopic detection of NSAID damage is all too frequently demonstrated after a severe complication (e.g., obstruction, perforation, or hemorrhage) becomes clinically apparent.
111In
Leucocytes Scan
The initial effect of NSAIDs to increase small intestine permeability is a prerequisite for the subsequent development of small intestine inflammation. This small intestinal inflammation induced by NSAIDs is associated with blood and protein loss, both of which may contribute to the general ill-health of rheumatic patients.[24,49,50] Further studies have shown that long-term NSAID treatment is associated with enhanced migration of indium 111-labeled leucocytes to the ileum indicating small bowel inflammation. Qualitative 111In-labeled leucocyte scintography is useful for determining the sites of affliction of the GI tract.[48][Figure 1]
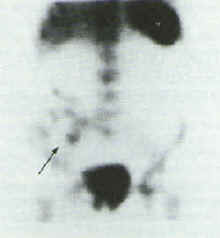
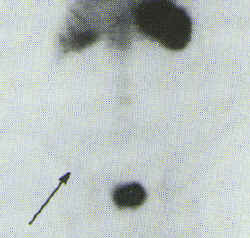
Figure 1. Left Panel 111In-labeled white blood cell scan, indicating an area of abnormal uptake in the small intestine (Arrow) Right Panel. Repeat 111In-labeled white blood cell scan 7 months after NSAID discontinuation, showing lack of uptake in the small bowel (arrow).[48] Adopted from reference [48] with permission.
111In
Faecal Counts
The 111In-labelled leucocytes scan together with a 4 day faecal collection measuring indium 111 excretion provides a sensitive quantitative measure of intestinal inflammatory activity.[46] These methods suggest that small intestinal inflammation occurs predominantly in the terminal jejunum and upper ileum.[46]
Faecal Calprotectin
Both pre-clinical and clinical studies have demonstrated that increased faecal calprotectin, a non-degraded neutrophil cytosolic protein, or granulocyte marker protein in rats are effective and sensitive markers for diagnosing NSAID enteropathy.[70-72]
Table 1. Intestinal Inflammation in Patients on Long-Term NSAIDs
|
|
No. |
No. |
4-day
fecal excretion of |
|
Normal Control |
22 |
0 |
0.5±0.2 |
|
Patient
Controls |
20 |
3 |
0.6±0.3 |
|
Aspirin |
7 |
1 |
0.7±0.3 |
|
Nabumetone |
13 |
2 |
1.1±0.4 |
|
Sulindac |
9 |
5 |
2.5±1.9* |
|
Fenbufen |
11 |
6 |
2.7±2.3* |
|
Ibuprofen |
29 |
16 |
3.0±1.2* |
|
Ketoprofen |
14 |
9 |
3.5±1.8* |
|
Etodolac |
11 |
7 |
3.7±2.1* |
|
Naproxen |
58 |
42 |
3.9±0.8* |
|
Piroxicam |
28 |
15 |
3.9±1.1* |
|
Diclofenac |
38 |
24 |
4.4±4.8* |
|
Flurbiprofen |
16 |
9 |
4.5±3.2* |
Values represent mean (SD) fecal excretion of Indium-111 labeled neutrophils (% dose) following intravenous instillation. * Differs significantly (ANOVA from normal and patient controls, p<0.05 [79]
The effect of short-term NSAID administration on faecal calprotectin shedding has been evaluated in two randomized crossover studies, with treatment regimens of indomethacin or naproxen for 14 days in the first study (n = 16), and lornoxicam or naproxen for 7 days in the second study (n = 18). The reproducibility and stability of quantitative faecal calprotectin sampling were satisfactory. Indomethacin and naproxen both significantly increased faecal calprotectin shedding from a base line of 4.7 mg/l to 9.0 mg/l and 8.0 mg/l respectively. Lornoxicam failed to increase faecal calprotectin shedding from baseline.[70] Single stool faecal calprotectin concentrations were compared to four day faecal excretion of 111In-labelled leukocytes to assess the prevalence and severity of NSAID enteropathy in 312 patients (192 with rheumatoid arthritis, 65 with osteoarthritis, and 55 with other conditions) taking 18 different NSAIDs.[71] The four day faecal excretion of 111In-labelled leukocytes was significantly correlated with faecal calprotectin concentrations. Faecal calprotectin concentrations were significantly higher in the patients on NSAIDs than in normal controls.[71] The prevalence and severity of protein shedding and leukocyte migration was independent of the particular type or dose of NSAID taken. Faecal calprotectin analysis seems to provide a simple, practical, and functional method for diagnosing NSAID-induced intestinal damage in man.[70,71]
Table 2. Fecal Calprotectin Concentration and Type of Non-steroidal Anti-inflammatory Drug Used
|
NSAID |
No.
Studied |
Calprotectin
(mg/ml) Median
(range) |
|
Normal
Controls |
48 |
2.0(0.2-10.9) |
|
Others |
13 |
4.0
(0.5-41.4) |
|
Meloxicam |
7 |
4.5(0.5-11.0)
† |
|
Nabumetone |
5 |
7.0(4.0-16.0)
† |
|
Etodolac
|
6 |
7.5(0.5-15.5)
† |
|
Piroxicam |
6 |
7.5(5.0-18.0)
† |
|
Ibuprofen |
35 |
7.0(1.0-61.0)* |
|
Indomethacin |
35 |
7.0(1.0-60.0)* |
|
Diclofenac
and misoprostol |
27 |
8.0(1.0-57.0)* |
|
Diclofenac |
116 |
8.0(1.0-118.0)* |
|
Naproxen |
62 |
9.0(1.0-60.0)* |
*Significantly
different from normals (p<0.001, Mann-Whitney U test).
†
Insufficient patient numbers for reliable statistical analysis.[69]
Small Intestinal Protein Loss
A small percentage of arthritic patients may have hypoalbuminaemia ascribed to protein-losing enteropathies independent of NSAIDs. To study the role of NSAIDs in protein loss Bjarnason, et al. simultaneously labeled leucocytes with 111indium and proteins with 51chromium. Using a 10 day fecal collection the correlation between faecal and serum 51Cr activities were made. Correlating enhanced 51Cr faecal to serum activity with increased ileal localization of 111In activity patients on long-term NSAID therapy demonstrated a protein losing entropathy.[49]
Small
Intestinal Bleeding
There
does not appear to be a close relationship between demonstrable lesions on
gastroduodenoscopy and gastrointestinal blood loss in NSAID-treated
patients.[73-75] Several approaches have been developed to evaluate
NSAID-induced bleeding in the more distal GI tract. Such distal blood loss
is an important contributing factor to anaemia in patients with rheumatoid
arthritis taking NSAIDs.[24,47,49-50,76] Approximately half of patients
with occult GI bleeding while on regular NSAIDs were found to have jejunal
or ileal ulcerations.[77]. NSAID-induced enteropathy appears to be
associated with low-grade blood loss which may be utilized as an early
diagnostic technique for NSAID-induced enteropathy in these patients.[3]
99mTc-Labeled
Erythrocytes
Following
NSAID ingestion in humans the simultaneous intravenous injection of
leucocytes labeled with 111indium and erythrocytes with 99m
technetium revealed identical congregations of both radioactive
blood cell types in the right iliac fossa. This experiment elegantly
correlated the inflammatory and haemorrhagic damage induced in the
intestinal tract with NSAID use.[3]
51Cr-EDTA
Labeled Erythrocytes
Bjarnason’s
group has also correlated intestinal inflammation with intestinal blood
loss. Intestinal inflammation was assessed with 111indium
leucocyte labeling. Small intestinal blood loss was assessed by labeling
erythrocytes with 51chromium- and correlating the 5 day fecal
excretion of the isotope with the previous' day's blood radioactivity
level. The fecal excretion of 111indium correlated
significantly with the mean daily intestinal blood loss (r = 0.59, p <
0.01).[49-50].
Prevalence of NSAID Enteropathy
Epidemiological
studies
The
prevalence of NSAID-associated ulcers in the gastroduodenum is
approximately 20%, and between 1 to 2% of patients chronically taking
NSAIDs suffer a serious bleeding or perforation complication per year of
NSAID use.[78] With substantial small intestinal toxicity and widespread
use the under-recognized morbidity from NSAIDs may be very substantial.
The prevalence of lower GI side-effects may exceed that detected in the
upper GI tract.[79] In
two retrospective, hospital-based, systematic studies of 268 and 188
respective patients hospitalized with intestinal perforations or
hemorrhage those patients on NSAIDs were twice as likely to have
complications develop compared to controls.[80]
Morris, et al. [47] reported that of 46 patients with rheumatoid
arthritis taking NSAIDs 41% had iron deficiency anemia with evidence of
small intestinal ulcers or erythema on enteroscopy.
A
recent report retrospectively examined the prevalence of surgical
complications from NSAID-associated small bowel ulcerations. Among 283
patients who underwent small bowel surgery there were 11 patients with 12
surgical complications undergoing emergent small bowel resections for
NSAID-induced small bowel ulcerations. Ulcers were confined to the jejunum
(4) and ileum (8), and were multiple in 50% of cases. Complications from
small bowel ulceration included bleeding (50%), perforation (33%) and
obstruction (17%) in the patients taking NSAIDs.[83]
Allison,
et al. [82] have recently reported an increased incidence of small
intestinal ulceration in patients prescribed NSAIDs, and, although these
ulcers are less common than those in the gastroduodenum, they still may be
life-threatening. Autopsies of 713 patients previously
taking NSAIDs identified jejunal or ileal ulcers in 8.4% compared to 0.6%
in controls. Long-term users of NSAIDs (> 6 months) were at a high risk
of developing ulcers (14%). In 3 patients (0.4%) death was attributed to
perforated small intestinal ulcers.
Clinical Surveillance with
Radiolabels
Several
studies have used radiolabels to survey patients for NSAID-induced
intestinal inflammation. In 97 patients on NSAIDs for > 2 months for
either rheumatoid arthritis or osteoarthritis 66% of patients had evidence
of NSAID enteropathy as detected by 111In scintigraphy scans
and stool collections.[24] Sigthorsson, et al. [81] demonstrated
that 54 to 72% of 286 patients on 12 different NSAIDs had evidence of
intestinal inflammation using 111In labeled leucocyte faecal
excretion studies.
In
312 patients taking NSAIDs for osteoarthritis or other rheumatic
conditions fecal calprotectin shedding correlated with 4 day 111In
fecal excretion (r=0.83). 44% of these patients were diagnosed with NSAID
enteropathy using this technique.[71]
Pathogenesis of NSAID-Induced
Intestinal Toxicity
NSAID-induced
damage to the intestinal epithelium results from three sources of
exposure: (1) pre-absorption
local effects following direct exposure after oral administration, (2)
systemic effects after absorption, and (3) the recurrent local effects
following enterohepatic recirculation. The relative damage inflicted from
each type of access remains unknown.
Traditionally,
the therapeutic and toxic effects of NSAIDs have been attributed to the
ability of these drugs to inhibit the synthesis of stable prostaglandins
by the direct inhibition of prostaglandin H synthetase, which serves both
as a cyclo-oxygenase and as a peroxidase.[84] Inhibition of
'cytoprotective' prostaglandin synthesis has been regarded as a major
factor in the development of gastrointestinal ulceration and
hemorrhage.[85] However, the lack of a specific link between inhibition of
prostaglandin synthesis and development of gastrointestinal damage is
exemplified by the absence of gastric lesions with reduced mucosal
concentrations of PGE2 and PGI2 after the
administration of various NSAIDs to rats.[85]
Cyclo-oxygenase
inhibition has also been implicated in causing the gastrointestinal damage
attributed to NSAIDs. Two subtypes of cyclo-oxygenase activity (COX-1 and
COX-2) have been identified. NSAIDs vary in their COX selectivity and
recent studies have demonstrated intriguing differences in COX-selective
toxicities. Lagenbach [86] has developed a transgenic knockout mouse
strain with homologous gene disruptions of COX-1 with resultant
deficiencies in the COX-1 isozyme activity. Surprisingly, these COX-1
deficient mice had no evident gastric or intestinal pathology, and
appeared less sensitive to NSAID-induced gastric ulceration. These
observations suggest that NSAID-induced epithelial damage may result from
mechanisms other than (or in addition to) COX-1 inhibition. These results
should be interpreted cautiously as these mice may have adapted
alternative defense mechanisms to overcome COX-1 deficiency, or that
secondary alterations in NSAID pharmacokinetics could have occurred.
Mielants, et al. [87] reported that there was no significant
difference in gut permeability between patients taking NSAID and patients
taking corticosteroids. This further suggests that alteration of gut
permeability may not solely be accounted for by an inhibition of
epithelial cyclo-oxygenase activity.
Animal
studies suggest that the pathogenesis of NSAID small intestinal toxicity
probably involves multiple interactions dependent on enterohepatic
recirculation, epithelial permeability, neutrophil infiltration, and
bacterial infection.[88-91] Increased intestinal permeability may allow
dietary macromolecules, bile acids, pancreatic juices, bacteria, and other
intra-luminal toxins access to the usually intact and protected intestinal
epithelium. The neutralization of such toxins is impaired by
NSAID-associated impairment of neutrophil function. Left in only partial
check by weakened host defences these toxins and infectious agents can
induce epithelial inflammation and subsequent fibrosis.[3] Submucosal
granulation tissue matures into collagenous scar tissue forming
contracting rings that acts as draw strings contracting the intestinal
lumen, the result is the characteristic 'diaphragm disease' associated
with NSAID enteropathy.[30].
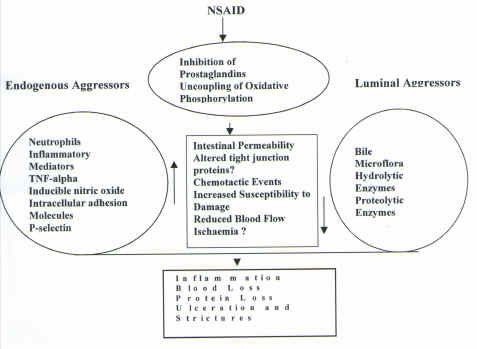
Schematic 1. Possible Pathogenesis of NSAID Induced Small Bowel Damage. (Adapted from references 3, 44 and Davies Unpublished Observations)
Treatment of NSAID-Induced
Enteropathy
As NSAIDs are definitely linked to the development of serious GI side-effects, numerous strategies have been employed to prevent or reduce this damage. The most effective means of reducing GI toxicity has been to withdraw the offending NSAID. This approach may not be clinically suitable for many patients dependent on NSAIDs for relief from crippling pain.
An alternative approach has been the concomitant use of protective substances to circumvent epithelial damage. The protective effects of sucralfate have been shown to be confined to the upper GI tract. Sucralfate did not provide protection from naproxen- induced permeability changes in the distal intestine.[113] Aabakken’s group have examined the use of the H2-antagonists cimetidine and famotidine, neither of which seem to possess any apparent protective or antagonizing effect on naproxen-induced intestinal permeability.[114-115]
NSAIDs, as COX inhibitors, decrease prostaglandin synthesis from fatty acids within intestinal cells.[3] Bjarnason and co-workers examined the influence of concomitant prostaglandin administration on indomethacin-induced permeability changes. Using prostin E2, a naturally-occurring prostaglandin, increased NSAID permeability was not reduced. Prostin E2 is unstable, and may have lost pharmacodynamic activity during the experiment.[116] High-dose misoprostol, a prostaglandin analogue, co-administered with indomethacin partially alleviated the indomethacin-induced increase in intestinal permeation of 51Cr-EDTA.[117-118] Rioprostol, another prostaglandin analogue, given in small doses at the same time as indomethacin also preserved intestinal integrity.[119] In contrast, ornoprostil, another prostaglandin E1 analogue, co-administered with indomethacin showed no significant change in intestinal permeability compared to controls.[98] Davies, et al. [120] showed only small reductions in intestinal permeability with misoprostol (800 µg) co-administration with indomethacin (150 mg). In Bjarnason’s reports [117-118] 1200 µg misoprostol was administered. In a small study (n = 6), misoprostol had no effect on reducing naproxen-induced increased intestinal permeability.[121] Neither the time course of naproxen-induced permeability changes nor the dose-dependency of misoprostol administration was considered. These studies suggest that prostaglandin alleviation of NSAID-induced intestinal permeability may be dose-dependent or that intestinal permeability may only be partially mediated by reduced mucosal prostaglandins. Morris, et al.[122] have retrospectively showed a small improvement in anemia in patients with rheumatoid arthritis on misoprostol therapy with endoscopically-proven NSAID enteropathy. Haemoglobin rose from a median of 9.1 g/dl to 10.6 g/dl with misoprostol therapy. Faecal calprotectin shedding remained unchanged despite the conversion from plain diclofenac to Arthrotec®, a combination of diclofenac and misoprostol. [71] [Table 2]
Table 3. Treatment of NSAID-Induced Permeability
|
Effective |
Reference |
|
Misoprostol |
117-118,
135 |
|
Rioprostol |
119 |
|
Ornoprostil |
98 |
|
Metronidazole |
120 |
|
Glutamine |
135 |
|
Sulphasalazine |
127 |
|
Glucose/citrate |
132 |
Table 4. Treatment of NSAID-Enteropathy
|
Effective |
Reference |
|
Misoprostol |
119 |
|
Metronidazole |
120 |
|
Sulphasalazine |
125-126 |
Co-administered metronidazole (400 mg twice daily for 7 days) significantly reduced indomethacin-induced permeability changes.[120] In another study, metronidazole (800 mg per day) was ineffective at reducing 51Cr-EDTA/rhamnose measured intestinal permeability, but did reduce 111In-labeled leucocyte measured inflammation and 51Cr labeled-erythrocyte measured haemorrhage.[123] Metronidazole is a bactericidal nitroimidazole antibiotic effective against many enteric anaerobic bacteria. These results support a two-stage pathogenesis of NSAID mucosal damage. Although metronidazole would not impede NSAID-induced increases in intestinal permeability, the load of inflammation-producing invading bacteria would be reduced.[120] Metronidazole is also used in the treatment of inflammatory bowel disease to treat active intestinal inflammation. Metronidazole undergoes reductive activation producing the nitro-free radical, nitroso, a nitroso-free radical, and hydroxylamine derivatives.[124] Metronidazole has also been shown to be a free radical scavenger.[125] Metronidazole also prevents both leucocyte adherence and transmigration.[126] It is also possible that the utility of metronidazole in NSAID enteropathy derives from its anti-adhesive effect rather than from its antibacterial activity.[126]
Sulfasalazine is a mainstay in the treatment of inflammatory bowel disease. Sulfasalazine (1 g twice daily for 3 days) also prevents acute indomethacin-induced increased intestinal permeability (75 mg orally twice daily for one day),[127], reduces faecal 111In excretion,[128] and reduces 51Cr-labeled erythrocyte blood loss.[129] This agent has anti-inflammatory effect through an adenosine-dependent mechanism,[130] and is speculated to inhibit adhesion molecule expression [131] thereby limiting neutrophil recruitment to sites of inflammation ameliorating further inflammatory process.
In addition to inhibiting prostaglandin synthesis, NSAIDs may inhibit glycolysis and the tricarboxylic acid cycle resulting in the inhibition of oxidative phosphorylation. Cell damage resulting from the depletion of adenosine triphosphate (ATP).[3] Collapse of the cytoskeleton follows ATP depletion, and intercellular junction integrity is compromised. Administering indomethacin with 15 mg glucose and 15 mg citrate for each milligram of indomethacin prevented an increase in intestinal permeability above baseline values.[132] It has been suggested that the presence of these sugars in the intestinal lumen may modify the reaction to indomethacin, or that citrate may be cytoprotective against the free radical damage caused by NSAIDs.[3,132] Unfortunately, no other studies have yet confirmed the protective effects of glucose or citrate. Glucose and citrate are ineffective following long-term repeated administration of indomethacin.[3]
Omega-3 fatty acid-rich fish oil ingestion can inhibit pro-inflammatory neutrophil leukotriene B4 and platelet activating factor release.[133] Fish oil supplementation was given to subjects on NSAIDs to assess possible protective effects from intestinal damage. In subjects taking indomethacin (50 mg thrice daily) the 51Cr-EDTA measured increase in intestinal permeability from baseline (2.7 ± 1.14%) was attenuated in subjects taking fish (2.34 ± 0.20%), but not corn (2.30 ± 0.99%) oil.[134]
As increased intestinal permeability might be related to cell damage resulting from energy depletion. It has been hypothesized that glutamine, a major energy source for the intestinal epithelium, might prevent NSAID-induced intestinal permeability changes. In 6 healthy volunteers pre-treatment with glutamine (7 g thrice daily 1 week before indomethacin 50 mg at 22:00 h prior to the permeability test, and at 09:00 on the day of testing) did not attenuate indomethacin-induced 51Cr-EDTA measured increases in permeability (0.42 ± 0.07%, 1.02 ± 0.23%, and 1.12 ± 0.22%, for baseline, indomethacin, and indomethacin plus glutamine respectively). A single dose of glutamine (7 g) taken a half hour before indomethacin in 12 volunteers did not influence permeability (0.78 ± 0.07%, 2.11 ± 0.34%, and 1.90 ± 0.27%, for baseline, indomethacin and indomethacin plus glutamine respectively). Multiple doses of glutamine before indomethacin dosing also did result in a significant reduction in intestinal permeability (0.56 ± 0.1%, 1.61 ± 0.21%, and 1.06 ± 0.13%, for baseline, indomethacin and indomethacin plus glutamine respectively). Misoprostol (400 mg on the day before and on the day of the test) reduced indomethacin-induced increases in intestinal permeability significantly (0.73 ± 0.08%, 1.71 ± 0.23%, and 1.28 ± 0.21% for baseline, indomethacin and indomethacin plus misoprostol respectively). In 11 volunteers co-administered glutamine and misoprostol further attenuation of indomethacin-induced increases in intestinal permeability occurred (0.63 ± 0.10%, 1.65 ± 0.19%, 1.266 ± 0.19%, and 0.87 ± 0.17% for baseline, indomethacin, indomethacin plus glutamine, and indomethacin plus glutamine plus misoprostol respectively).[135]
Do
Pro-Drugs Spare the Intestinal Tract?
Some
NSAIDs are formulated as pro-drugs that are inactive as COX inhibitors
until after post-absorption metabolism. Nabumetone is a pro-drug for its
6-methoxynapthylacetic (6-MNA) acid metabolite. Neither nabumetone nor its
6-MNA metabolite are excreted in bile avoiding the possibility of
enterohepatic recirculation.[92] Following the administration of 1 g of
nabumetone daily for seven days to 12 healthy humans 51Cr-EDTA
measured permeability remained at baseline. In contrast indomethacin, an
active COX inhibitor with enterohepatic recirculation, significantly
enhanced 51Cr-EDTA measured permeability.[93] Several subjects
taking nabumetone did, however, show statistically non-significant trends
towards increased 51Cr-EDTA measured permeability in this small
study. A recent abstract also suggested nabumetone has the potential to
increase in intestinal permeability in healthy volunteers.[94] A
follow up study of 13 patients demonstrated less 111In faecal
excretion in patients administered nabumetone compared to other NSAIDs.
Nabumetone-treated patients had similar 111In faecal excretion
to control patients and healthy volunteers not taking NSAIDs (1.1±0.4,
0.6±0.3
and 0.5±0.2%,
respectively, p NS).[81] Only one (16 mg/L) of five patients with chronic
arthropathies taking nabumetone had elevated faecal calprotectin
concentrations compared to 48 controls (median 2.0 mg/L, range 2.0 to 10.9
mg/L).[71] Nabumetone may decrease NSAID-induced gastrointestinal damage,
however, further and larger trials are required to confirm this
preliminary observation. Nabumetone is not without gastrointestinal
side-effects, and increased clinical vigilance should be maintained when
patients are undergoing therapy with this NSAID.[95]
Sulindac,
a pro-drug, is a metabolised to sulindac sulphide and undergoes minimal
enterohepatic recirculation (4%).[96] Sulindac (200 mg daily for 7 days)
did not appear to induce an increase in 51Cr-EDTA permeability
above baseline when compared to indomethacin (2 mg/kg/day).[97] 51Cr-EDTA
measured permeability (1.15 ± 0.89 vs 0.61±0.26% respectively) and
faecal 111In excretion (2.5 ± 1.9 vs 0.6±0.3% respectively)
did increase in 9 patients with rheumatoid arthritis on sulindac (300 to
400 mg/day) compared to controls.[81] Two case reports have suggested an
association between sulindac and small bowel damage.[23,41]
In
11 healthy volunteers indomethacin (75 mg over 1 day), but not its
pro-drug, acematacin, significantly increased intestinal permeability as
measured by lactulose/rhamnose excretion in urine.[98]
Acetylsalicylic
acid (ASA) is metabolized into salicylic acid. In two studies (total n =
21) there has been a trend for ASA not to increase small intestinal
permeability.[79,81] Conversely, in a third study ASA increased intestinal
permeability in 13 of 16 (81%) of patients.[99] Other case reports
corroborate an ASA-induced enteropathy and associated intestinal
bleeding.[67,73,76]
Enteric-Coated
and Slow-Release NSAID Formulations
Enteric coating of NSAIDs has been attempted to allow the drug to bypass the gastroduodenum before dissolving thereby diminishing gastroduodenal exposure to the active drug. Such enteric coating of several NSAIDs reduced endoscopic lesions in the stomach and duodenal bulb.[100-101]. These formulations can increase the exposure and toxicity of active drug to the more distal intestine. A controlled-release indomethacin preparation, Osmosin®, was suspected to cause intestinal perforation, and it was voluntary removed from the market.[102-104] Several case reports associate the use of enteric-coated and sustained-release NSAID formulations with small bowel toxicity.[105] A latin-square crossover study of healthy men given naproxen 500 mg twice daily for 7 days either as plain tablets, enteric-coated tablets, or encapsulated enteric-coated granules showed statistically similar increases in 51Cr-EDTA measured permeability for all formulations. Considerable inter-individual variation was seen with greater median urinary excretion values for the coated tablets and encapsulated coated granules than for the plain tablets.[106] Other investigators have reported increased intestinal permeability with a sustained-release but not regular formulation of diclofenac.[107] Available data suggest that sustained release NSAID formulations do not solve the problem of NSAID-induced enteropathy.
Selective COX-2 Inhibitors
As expected, the discovery of COX isozymes has prompted many investigators to search for molecules effective in inhibiting the inducible COX-2 isozyme with little or no effect on constitutive COX-1 isozyme. As NSAID-induced epithelial damage may be partially mediated through prostaglandin-independent mechanisms it is unknown if this suggested strategy will alleviate intestinal lesions. Pre-clinical studies of highly selective COX-2 inhibitors in animal models suggest that these agents may reduce gastric toxicity compared to non-selective NSAIDs.[136-139] Nimesulide, a COX-2 selective NSAID (200 mg per day for 10 days), was been compared with naproxen, a COX non-selective NSAID (500 mg twice daily for 10 days) in 23 subjects surveyed with intestinal permeability and faecal calprotectin tests. Nimesulide did not increase intestinal permeability or calprotectin shedding, whereas naproxen increased both markers of NSAID damage.[140]
Rofecoxib (VioxxÒ), a selective COX-2 inhibitor, was studied in a 4-way crossover study after taking drugs for a week with a subsequent week-long washout period. Placebo was compared to indomethacin (50 mg taken thrice) and rofecoxib (25 mg and then 50 mg with one dose of a placebo for blinding). Only the indomethacin treatment produced significantly higher lactulose/L-rhamnose permeability ratios (0.58 vs 0.34 to 0.36 for the other treatments). No subject developed increased intestinal permeability with rofecoxib.[141, and Bjarnason, personal communication].
Meloxicam is another new preferential COX-2 inhibitor with a favorable pharmacokinetic and tolerability profile.[142] In two large comparative prospective trials (MELISSA and SELECT) meloxicam had significantly less gastrointestinal toxicity than non-selective NSAIDs in osteoarthritis patients.[143-144] The rat has previously been demonstrated to be a good model for NSAID-induced small intestinal toxicity.[145-149] Meloxicam, celecoxib and indomethacin were administered in therapeutically equivalent doses to rats. Meloxicam and celecoxib both demonstrated insignificant amount of intestinal damage compared to indomethacin which produced a striking and profuse enteropathy.[Figure 2]
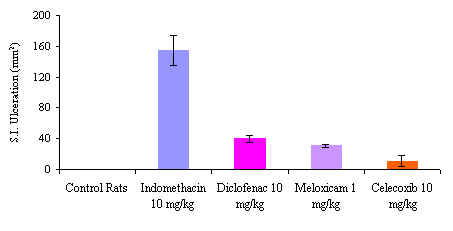
Figure 2. Small Intestinal Ulveration after NSAIDs in the Rat Model N=4, Mean± S.E.M
One short-term study has evaluated
gastric and intestinal permeability following the ingestion of
slow-release formulated indomethacin and plain meloxicam for 2 days. No
significant increases in gastroduodenal permeability were reported with
either drug. 70% of subjects taking meloxicam had normal intestinal
permeability, and mean recovered lactulose/mannitol ratios showed
insignificant increases following either indomethacin or meloxicam
administration (0.021 ±
0.01, 0.032 ±
0.03 and 0.035 ±
0.02 for controls, meloxicam or indomethacin respectively). In 7 patients
meloxicam did not increase intestinal inflammation as assessed by fecal
calprotectin shedding compared to control patients.[71] Short-term
permeability studies and pre-clinical evaluation in the rat may have
predictive value for longer-term outcome with respect to meloxicam
enteropathy.[Table 2]
Celecoxib, a COX-2 selective NSAID,
may also have intestinal sparing properties although definitive human
trials are eagerly awaited. Other selective COX-2 inhibitors (ie.
valdecoxib, parecoxib and deracoxib) are currently in development. There
is little published information on relative intestinal toxicities of
COX-selective NSAIDs. Celecoxib has an excellent gastric tolerability
profile since its introduction in 1999. There have, however, been cases of gastrointestinal hemorrhages with celecoxib since its
introduction in the United States with at least five deaths potentially
from GI bleeding.[150-151]
Experimental
Approaches
There have been numerous experimental approaches evaluated over the years that have shown promise in pre-clinical studies in reducing the problem of NSAID enteropathy. NO-NSAIDs are NSAIDs with a nitric oxide moiety linked to the carboxylic ester functional group. Since 1993, numerous pre-clinical studies have demonstrated reduced gastrointestinal side-effects of these drugs.[152-153] These compounds are currently in clinical trials, however, there are no published clinical studies available in the literature. Interestingly, some pre-clinical data suggests that NO-NSAIDs demonstrate markedly reduced bioavailability compared to the parent NSAIDs.[72, 153] The formulating of NSAIDs with zwitterionic phospholipids has also demonstrated pre-clinical efficacy with reduced gastrointestinal toxicity.[154] These compounds are also in clinical trials and there is one published clinical study demonstrating reduced gastric toxicity.[155] Lastly, an intriguing approach to reduce gastroenteropathy is the formulation of NSAIDs with metallic coordination compounds.[156-157] These NSAID coordination complexes [Figure 4 and 5] already in wide-spread veterinary use in Australia [Figure 6] and undergoing clinical trials appear to have negligible NSAID gastroenteropathy or renal toxicity while maintaining anti-inflammatory efficacy.[156] However, in all three of these novel approaches more clinical data and more thourough pharmacokinetic investigations of these compounds must be undertaken and ascertained. Further clinical studies of each of these approaches which may potentially solve difficult and serious clinical problems are eagerly awaited.
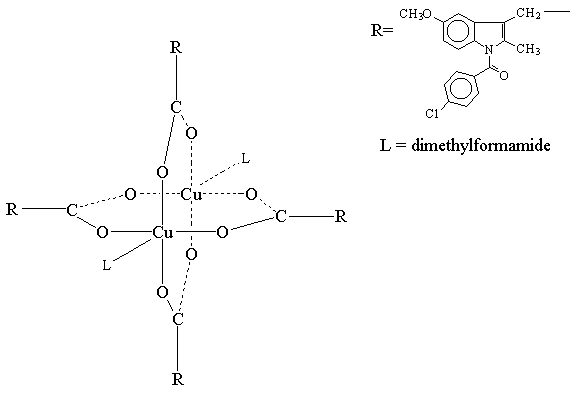
Figure 4. Crystal Stucture of Copper Indomethacin
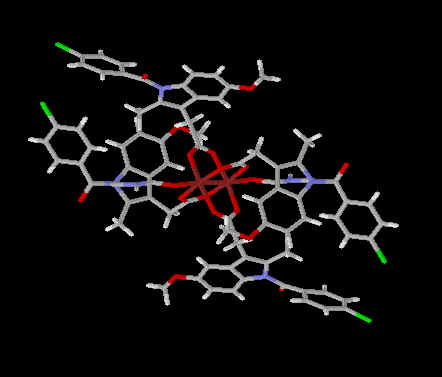
Figure 5. Three Dimensional Structure of Copper Indomethacin
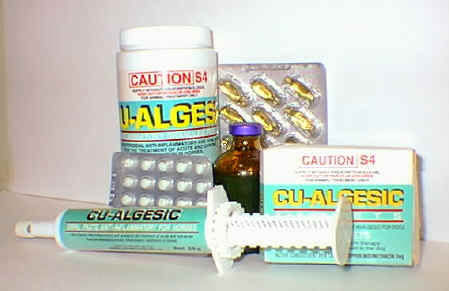
Figure 6. Veterinary formulations of copper indomethacin currently marketed in Australia.
Conclusions
NSAID enteropathy exists and may have clinically significant sequelae. Increased awareness of this potential avoidable condition is required for early recognition and appropriate management. The association between NSAID therapy and small bowel inflammation is worth emphasizing since unnecessary and protracted investigations can be avoided in some patients if this association is appreciated. COX isozyme non-selective NSAIDs induce a broad spectrum of gastrointestinal side effects throughout the GI tract reflecting their widespread use and high toxicity. The forthcoming generation of selective and preferential COX-2 inhibitors, such as meloxicam, celecoxib, rofecoxib and nimesulide, offer a potential therapeutic advance in terms of reducing side-effects throughout the gastrointestinal tract. The results of other experimental approaches are eagerly awaited.
References
-
Heller CA, Ingelfinger JA, Goldman P. Nonsteroidal anti-inflammatory drugs and aspirin: analyzing the scores. Pharmacotherapy 1985; 5: 30-8.
-
Walker JS, Sheather-Reid RB, Carmody JJ, Vial JH, Day RO. Nonsteroidal anti-inflammatory drugs in rheumatoid arthritis: support for the concept of “responders” and “non-responders”. Arth Rheum 1997; 40: 1944-51.
-
Bjarnason I, Hayllar J, Macpherson AJ, Russell AS. Side effects of nonsteroidal anti-inflammatory drugs on the small and large intestine in humans. Gastroenterology 1993; 104:1832-47.
-
Myers ABR. Salicin in acute rheumatism. Lancet 1876; 2: 676-677.
-
Kent TH, Cardelli RM, Stamler FW. Small intestinal ulcers and intestinal flora in rats given indomethacin. Am J Pathol. 1969; 54: 237-245.
-
Upadhyay R, Torley HI, McKinlay AW, Sturrock RD, Russell RI. Iron deficiency anaemia in patients with rheumatic disease receiving non-steroidal anti-inflammatory drugs: the role of upper gastrointestinal lesions. Ann Rheum Dis 1990; 49:359-62.
-
Silvoso G, Ivey KJ, Butt J. Incidence of gastric lesions in patients with rheumatic disease on chronic aspirin therapy. Ann Intern Med 1979; 91: 517-520.
-
Lennert VKA, Kootz F. Nil nocre! Arzneimittelbedingte dünndardmulzera. Münch. Med Wochenschr 1967; 40: 2058-2062.
-
Davies DR, Brightmore T. Idiopathic and drug-induced ulceration of the small intestine. Brit J Surg 1970; 57: 134-139.
-
Sturges HF, Krone CL. Ulceration and stricture of the jejunum in a patient on long-term indomethacin therapy. Am J Gastroenterol 1973; 59: 162-169.
-
Chadwick RG, Hossenbocus A, Colin-Jones DG. Steatorrhea complicating therapy with mefenamic acid. Br Med J 1976; 1(6006): 397.
-
Nagaraj HS, Sandhu AS, Cook LN, Buchino JT, Groff, D. Gastrointestinal perforation following indomethacin therapy in low birth weight infants. J Pediatr Surgery 1981; 166: 1003-1007.
-
Marlow N, Chiswick ML. Neonatal bartter’s syndrome, indomethacin and necrotising enterocolitis. Acta Paediatr Scand 1982; 71: 1031-1032.
-
Neoptolemos JP, Locke TJ. Recurrent small bowel obstruction associated with phenylbutazone. Br J Surg 1983; 70: 244-245.
-
Hall RI, Pelty AH, Cobden I, Lendrum R. Enteritis and colitis associated with mefenamic acid. Br Med J 1983; 21: 287: 1182.
-
Alpan G, Eyal F, Vinograd I, Udassin R, Amir G, Mogle P, Glick B. Localized intestinal perforation after enteral administration of indomethacin in premature infants. J Pediatr 1985; 106: 277-281.
-
Kühl G, Willie L, Bolkenius M, Seyburth HW. Intestinal perforation associated with indomethacin treatment in premature infants. Eur J Pediatr 1985; 143: 213-216.
-
Stewart JT, Pennington CR, Pringle R. Anti-inflammatory drugs and bowel perforations and haemorrhage. BMJ 1985; 290: 787-788.
-
Hervas JA, Masip MC, Alomar A, Bergante JI. Localized intestinal perforation after intravenous indomethacin in a premature infant. Helv Paediat Acta 1986; 41: 437-440.
-
Madok R, Mackenzie JA, Lee FD, Bruckner FE, Terry TR Sturrock RD. Small bowel ulceration in patients receiving NSAIDs for rheumatoid arthritis. Q J Med 1986; 58: 53-58.
-
Saverymuttu SH, Thomas, A, Grundy A, Maxwell J.D. Ileal stricturing after long-term indomethacin treatment. Postgrad Med J 1986; 62: 967-968.
-
Saw KC, Quick CRG, Higgins AF. Ileocaecal perforation and bleeding - are non-steroidal anti-inflammatory drugs (NSAIDs) responsible? J R Soc Med 1990; 83: 114-115.
-
Freeman HJ. Sulindac-associated small bowel lesion. J Clin Gastroenterol 1986; 8: 569-571.
-
Bjarnason I, Zanelli G, Smith T, Prouse P, Williams P, Smethurst P, Delacey G, Gumpel MJ, Levi AJ. Nonsteroidal antiinflammatory drug-induced intestinal inflammation in humans. Gastroenterology 1987; 93: 480-489.
-
Johnston F. Recurrent small bowel obstruction associated with piroxicam. Br J Surg 1987; 11: 74:654.
-
Isaacs PET, Sladen GE, Filipe I. Mefenamic acid enteropathy. J Clin Pathol 1987; 40: 1221-1227.
-
Sukumar L. Recurrent small bowel obstruction associated with piroxicam. Br J Surg 1987; 74: 186.
-
Bem JL, Mann RD, Coulson R. Fatal gastrointestinal damage associated with the use of osmotic mini pump indomethacin (Osmosin). Pharmaceut Med 1988; 3: 35-43.
-
Bjarnason I, Price AB, Zanelli G, Smethurst P, Burke M, Gumpel M, Levi AJ. Clinicopathological features of nonsteroidal antiinflammatory drug-induced small intestinal strictures. Gastroenterology 1988; 94: 1070-1074.
-
Lang J, Price AB, Levi A J, Burk M, Gumpel MJ, Bjarnason I. Diaphragm disease: the pathology of non-steroidal anti-inflammatory drug induced small intestinal strictures. J Clin Pathol 1988; 41: 516-526.
-
Levi S, de-Lacey G, Price AB, Gumpel MJ, Levi AJ, Bjarnason I.. 'Diaphragm-like' strictures of the small bowel in patients treated with non-steroidal anti-inflammatory drugs. Br J Radiol 1990; 63: 186-189.
-
McCune KH, Allen D, Cranley B. Small bowel diaphragm disease - strictures asscoiated with non-steroidal anti-inflammatory drugs. Ulster Med J 1992; 61: 182-184.
-
Hershfield NB. Endoscopic demonstration of non-steroidal anti-inflmammatory drug-induced small intestinal strictures. Gastroint Endosc 1992; 38: 388-390.
-
Matsuhashi N, Yamada A, Hiraishi M, Konishi T, Minota S, Saito T, Sugano K, Yazaki Y, Mori M, Shiga J. Multiple strictures of the small intestine after long-term nonsteroidal anti-inflammatory drug therapy. Am J Gastroent 1993; 87: 1183-1186.
-
Keating JP, Rees M. Diaphragm disease: a rare cause of small-bowel obstruction. Hospital Update 1992; 1: 48-49.
-
Keating JP, McIlwaine J. Simultaneous small and large bowel ulceration associated with short term NSAID use. NZ Med J 1993; 106 (965): 438.
-
Fellows IW, Clarke JMF, Roberts PF. Non-steroidal anti-inflammatory drug-induced jejunal and colonic diaphragm disease: a report of two cases. Gut 1992; 33: 1424-1426.
-
Going J, Canvin J, Sturrock R. Possible precursor of diaphragm disease in the small intestine. Lancet 1993; 341: 638-639.
-
Kwo PY, Tremaine WJ. Nonsteroidal anti-inflammatory drug-induced enteropathy: case discussion and review of the literature. Mayo Clin Proc 1995; 70: 55-61.
-
Zalev AH, Gardiner GW, Warren RE. NSAID injury to the small intestine. Abdom Imaging 1998; 23: 40-4.
-
Yoon KH, Ng SC. A case of sulindac-induced enteropathy resulting in jejunal perforation. Ann Acad Med Singapore 1998; 27: 116-9.
-
Giacoia GP, Azubuike K, Taylor JR. Indomethacin and recurrent ileal perforations in a preterm infant. J Perinatol 1993; 13:297-9.
-
Aabakken L. Non-steroidal anti-inflammatory drugs - the extending scope of gastrointestinal side effects. Aliment Pharmacol Ther 1992; 6: 143-162.
-
Aabakken L. Small-bowel side-effects of non-steroidal anti-inflammatory drugs. Eur J Gastroenterol Hepatol 1999; 11:383-8.
-
Morris AJ. Nonsteroidal anti-inflammatory drug enteropathy. Gastrointest Endosc Clin N Am 1999; 9:125-33.
-
Bjarnason I, Macpherson A. The changing gastrointestinal side effect profile of non-steroidal anti-inflammatory drugs. A new approach for the prevention of a new problem. Scand J Gastroenterol Suppl 1989; 163:56-64.
-
Morris AJ, Wasson LA, Mackenzie JF. Small bowel enteroscopy in undiagnosed gastrointestinal blood loss Gut 1992; 887-889.
-
Davies NM, Jamali F, Skeith KJ. Nonsteroidal antiinflammatory drug(NSAID)-Induced Enteropathy and Severe Chronic Anemia in a Patient with Rheumatoid Arthritis. Arth Rheum 1996; 39: 321-324.
-
Bjarnason I, Prouse P, Smith T, Gumpel MJ, Zanelli G, Smethurst P, Levi S, Levi AJ. Blood and protein loss via small intestinal inflammation induced by non-steroidal anti-inflammatory drugs. Lancet 1987; 2: 711-714.
-
Bjarnason I, Zanelli G, Smith T, Smethurst P, Price AB, Gumpel MJ, Levi AJ. The pathogenesis and consequence of non-steroidal anti-inflammatory drug induced small intestinal inflammation. Scand J Rheumatol Suppl 1987; 64: 55-62.
-
Bjarnason I, So A, Levi AJ, Peters TJ, Williams D, Zanelli GD, Gumpel JM, Ansell B. Intestinal permeability and inflammation in rheumatoid arthritis: Effects of non-steroidal anti-inflammatory drugs. Lancet 1984; 2: 1171-1173.
-
Rooney PJ, Jenkins RT, Smith KM, Coates G. 111Indium-labelled polymorphonuclear leucocyte scans in rheumatoid arthritis-an important clinical cause of false positive results. Br J Rheumatol 1986; 25: 167-170.
-
Aguirre Placio A, Romero Gomez M, Grilo Reina A, Rafel Ribas E. [An ileal ulcer and diaphragm-type colonic stenosis due to diclofenac]. Gastroenterol Hepatol 1999; 22: 232-4.
-
Gaede PH, Helmsoe-Zinck L, Brynskov J [Diaphragm-like strictures of the small intestine after treatment with non-steroidal anti-inflammatory agents]. Ugeskr Laeger 1993; 155: 2409-11
-
Deakin M. Small bowel perforation associated with an excessive dose of slow release diclofenac sodium. Brit Med J 1988; 297: 488-489.
-
Bjarnason I, Peters TJ. Intestinal permeability, non-steroidal anti-inflammatory drug enteropathy and inflammatory bowel disease: an overview. Gut 1989; 30: 22-28
-
Meddings JB, Sutherland LR, Byles NI, Wallace JL. Sucrose: A novel permeability marker for gastroduodenal disease. Gastroenterology 1993; 104: 1619-1626.
-
Aabakken L. 51Cr-Ethylenediaminetetraacetic acid absorption test: methodological aspects. Scand J Gastroenterol 1989; 24: 351-358.
-
Davies NM. Non-steroidal anti-inflammatory drug-induced gastrointestinal permeability. Aliment Pharmacol Ther 1998; 12: 303-20.
-
Bjarnason I, Peters TJ, Levi AJ. Intestinal permeability: Clinical correlates. Dig Dis 1986; 4: 83-92.
-
Bjarnason I, Peters TJ, Veall N. A persistent defect in intestinal permeability in coeliac disease demonstrated by a 51Cr-labeled EDTA absorption test. Lancet 1983; 1: 323-325.
-
Bjarnason I, O'Morain C, Levi AJ, Peters TJ. Absorption of 51Chromium-labeled ethylenediaminetetraacetate in inflammatory bowel disease. Gastroenterology 1983; 85: 318-22.
-
Simpson LO. NSAID and the leaky gut. Lancet 1985; 1: 218-219.
-
Aabakken L, Osnes M. 51Cr-Ethylenediaminetetraacetic acid absorption test. Effects of naproxen, a non-steroidal, antiinflammatory drug. Scand J Gastroenterol 1990; 25: 917-924.
-
Elia M, Behrens R, Northrop C, Wraight P, Neale G. Evaluation of mannitol, lactulose and 51Cr-labeled ethylenediaminetetra-acetate as markers of intestinal permeability in man. Clin Sci 1987; 73: 197-204.
-
Jenkins AP, Trew DR, Crump BJ, Nukajam WS, Foley JA, Menzies IS, Creamer B. Do non-steroidal anti-inflammatory drugs increase colonic permeability? Gut 1991; 32: 66-69.
-
Cutler C, Rex DK, Cummings OW. Per anus enteroscopic demonstration of a non-steroidal anti-inflammatory drug-induced ileal stricture. Gastroint. Endos. 1993; 39: 601-603.
-
Achanta KK, Petros JG, Cave DR, Zinny M. Use of intraoperative enteroscopy to diagnose nonsteroidal anti-inflammatory drug injury to the small intestine. Gastrointest Endosc 1999; 49: 544-6
-
Davies GR, Benson MJ, Gertner DJ, Van Someren RM, Rampton DS, Swain CP. Diagnostic and therapeutic push type enteroscopy in clinical use. Gut 1995; 37: 346-52.
-
Meling TR, Aabakken L, Roseth A, Osnes M. Faecal calprotectin shedding after short-term treatment with non-steroidal anti-inflammatory drugs. Scand J Gastroeneterol 1996; 31: 339-344.
-
Tibble JA, Sigthorsson G, Foster R, Scott D, Fagerhol MK, Roseth A, Bjarnason I. High prevalence of NSAID enteropathy as shown by a simple faecal test. Gut 1999; 45: 362-366.
-
Davies NM, Roseth AG, Appleyard CB, McKnight W, Del Soldato P, Calignano A, Cirino G, Wallace JL. No-naproxen vs. naproxen: ulcerogenic, analgesic and anti-inflammatory effects. Aliment Pharmacol Ther 1997; 11: 69-79.
-
Holt S, Rigoglioso V, Sidhu M, Irshad M, Howden CW, Mainero M. Nonsteroidal antiinflammatory drugs and lower gastrointestinal bleeding. Dig Dis Sci 1993; 38: 1619-1623.
-
Hedenbro Jl, Wetterberg P, Vallengren S, Bergquist L. Lack of correlation between fecal blood loss and drug-induced gastric mucosal lesions. Gastrointes Endosc 1988; 34: 247-51.
-
Collins AJ, Du Toit JA. Upper gastrointestinal findings and faecal occult blood in patients with rhumatic diseases taking nonsteroidal anti-inflmmatory drugs. Br J Rheumatol 1987; 26: 295-8.
-
Lanas A, Sekar C, Hirschowitz BI. Objective evidence of aspirin use in both ulcer amd nonulcer upper and lower gastrointestinal bleeding. Gastroenterology 1992; 103: 862-869.
-
Morris AJ, Madhok R, Sturrock RD, Capell HA, MacKenzie JF. Enteroscopic diagnosis of small bowel ulceration in patients receiving non-steroidal anti-inflammatory drugs. Lancet 1991; 337: 520.
-
Giercksky KE. Mucosal protection by H2 antagonists against injury by non-steroidal anti-inflammatory agents. Scand J Gastroenterol Suppl 1989; 163: 32-35.
-
Bjarnason I, Zanelli G, Prouse P, Williams P, Gumpel MJ, Levi AJ. Effect of non-steroidal anti-inflammatory drugs on the human small intestine. Drugs 1986; 32 (Suppl): 35-41.
-
Langman MJS, Morgan L, Worall A. Use of inflammatory drugs by patients admitted with small or large bowel perforations and haemorrhage. Br Med J 1985; 290: 347-349.
-
Sigthorsson G, Tibble J, Hayllar J, Menzies I, Macpherson A, Moots R, Scott D, Gumpel MJ, Bjarnason I. Intestinal permeability and inflammation in patients on NSAIDs. Gut 1998; 43: 506-11.
-
Allison MC, Howatson AG, Torrance CJ, Lee FD, Russell RI. Gastrointestinal damage associated with the use of nonsteroidal antiinflammatory drugs. N Engl J Med. 1992; 327: 751-756.
-
Kessler WF, Shires GT 3rd, Fahey TJ 3rd. Surgical complications of nonsteroidal antiinflammatory drug-induced small bowel ulceration. J Am Coll Surg 1997; 185(3): 250-4.
-
Whittle BJR. Temporal relationship between cycloxygenase inhibition, as measured by prostacyclin biosynthesis, and the gastrointestinal damage induced by indomethacin in the rat. Gastroenterology 1981; 80: 94-98.
-
Whittle BJR, Vane JR. A biochemical basis for the gastrointestinal toxicity of non-steroid anti-rheumatoid drugs. Arch Toxicol 1984; (suppl) 7: 315-322.
-
Langenbach R, Morham SG, Tiano HF, Loftin CD, Ghanayem BI, Chluda PC, Mahler JF, Lee CA, Goulding EH, Kluckman KD, Kim HS, Smithies O. Prostaglandin synthase 1 gene disruption in mice reduces arachidonic acid-induced inflammation and indomethacin-induced gastric ulceration. Cell 1995; 83: 483-492.
-
Mielants H, Goemaere S, De Vos M, Schelstraete K, Goethal K, Maertens M, Ackerman C, Veys EM. Intestinal mucosal permeability in inflammatory rheumatic diseases. I. Role of antiinflammatory drugs. J Rheumatol.1991;18: 389-393.
-
Reuter BK, Davies NM, Wallace JL. Nonsteroidal anti-inflammatory drug enteroapthy in rats: role of permeability, bacteria, and enterohepatic recirculation. Gastroenterology 1997; 112: 109-117.
-
Robert A, Asano T. Resistance of germfree rats to indomethacin-induced intestinal lesions. Prostaglandins 1977; 14: 333-341.
-
Wax J, Clinger WA, Varner P, Bass P, Winder CV. Relationship of the enterohepatic cycle to ulcerognesis in the rat small bowel with flufenamic acid. Gastroenterology 1970; 58: 772-780.
-
Miura S, Suematsu M, Tanaka S. Microcirculatory disturbance in indomethacin-induced intestinal ulcer. Am J Physiol 1991; 261: G213-G219.
-
Brett MA, Buscher G, Ellrich E, Greb WH, Kurth HJ, Rulander G, Schmerenbeck B, Haddock RE, Thawley AR. Nabumetone evidence for the lack of enterohepatic circulation of the active metabolite 6-MNA in humans. Drugs 1990; 40 (Suppl. 5): 67-70.
-
Bjarnason I, Fehilly B, Smethurst P, Menzies IS, Levi AJ. Importance of local versus systemic effects of non-steroidal anti-inflammatory drugs in increasing small intestinal permeability in man. Gut 1991; 32: 275-7.
-
Devlin J, Moots R, Andrews D, Boivin C, Emery P. High intestinal permeability in patients with inflammatory arthritis is more strongly associated with any NSAID therapy than with diagnosis or disease activity. Arth Rheum 1996; S-280 Abstract 1513.
-
Voss GD. GI bleeding associated with nabumetone. Am J Hosp Pharm 1994; 51: 2506, 2508.
-
Dujovone CA, Pitterman A, Vincek WC, Drorinska MR, Davies RO, Duggan DE Enterohepatic circulation of sulindac and metabolites. Clin Pharmacol Therap 1983; 33: 172-177.
-
Davies GR, Rampton DS. The pro-drug sulindac may reduce the risk of intestinal damage associated with the use of conventional non-steroidal anti-inflammatory drugs. Aliment Pharmacol Therap 1991; 5: 593-598.
-
Nagase K, Hiwatashi N, Ito K, Maekawa H, Noguchi M, Kinouchi Y, Toyota T. [Effects of NSAIDs and PGE1 analogue on the permeability of human small intestine].[Article in Japanese] Nippon Shokakibyo Gakkai Zasshi 1997;94:469-74.
-
Jenkins RT, Rooney RT, Jones DR, Bienenstock J, Goodacre RL. Increased intestinal permeability in patients with rheumatoid arthritis: a side-effect of oral non-steroidal antiinflammatory drug therapy? Br J Rheumatol 1987; 26: 103-7.
-
Lanza FL, Royer GL, Nelson RS. Endoscopic evaluation of the effects of aspirin, buffered aspirin, and enteric-coated aspirin on gastric and duodenal mucosa. N Engl J Med 1980; 303: 136-138.
-
Trondstad RI, Aadland E, Holler T, Olaussen B. Gastroscopic findings after treatment with enteric-coated and plain naproxen tablets in healthy subjects. Scand J Gastroenterol 1987; 20: 239-242.
-
Day TK. Intestinal perforation associated with osmotic slow release indomethacin capsules. Br Med J 1983; 287: 1672-1672.
-
Cree, IA, Walker, MA, Wright, M, Forrester JS. Osmosin and ileal ulceration: a case report. Scot Med J 1985; 30: 40-41.
-
Laidler P, Maslin SC, Gilhome RW. What's new in Osmosin and intestinal perforation? Path Res Pract 1985; 180: 74-76.
-
Davies NM. Sustained release and enteric coated NSAIDs: are they really GI safe? J Pharm Pharmaceut Sci 1999; 2: 130-139.
-
Aabakken L, Bjørnbeth BA, Hofstad B, Olaussen B, Larsen S, Osnes M. Comparison of the gastrointestinal side effects of naproxen formulated as plain tablets, enteric-coated tablets, or enteric-coated granules in capsules. Scand J Gastroenterol 1989; 24: (Suppl. 163), 65-73.
-
Choi VMF, Coates JE, Chooi J, Thomson ABR, Russell AS. Small bowel permeability - a variable effect of NSAIDs. Clin Invest Med 1995; 18: 357-361.
-
Smith MD, Brooks PM. Increased bowel permeability in ankylosing spondylitis and active RA. Aust NZ J Med 1984; 14 (suppl 1): 345-46.
-
Sundquist T, Magnusson KE, Sjödahl R, Stjerndtröm I, Tagesson C. Passage of molecules through the wall of the gastrointestinal tract. II. Application of low-molecular weight polyethylene glycol and a deterministic mathematical model for determining intestinal permeability in man. Gut 1980; 21: 208-214.
-
Juby LD, Rithwell J, Axon ATR. Lactulose / mannitol test: an ideal screen for celiac disease. Gastroenterology 1989; 96: 79-85.
-
O'Mahoney S, Ferguson A. Small intestinal mucosal protection mechanisms and their importance in rheumatology. Ann Rheum Dis 1991;. 50: 331-336.
-
Oman H, Henriksson AE, Johansson SG, Blomquist L. Detection of naproxen-induced intestinal permeability change may be facilitated by adding a standardized meal but not by forming marker ratios. Scand J Gastroenterol 1996;31:1182-8
-
Aabakken L, Larsen S, Osnes M. Sucralfate for prevention of naproxen-induced mucosal lesions in the proximal and distal gastrointestinal tract. Scand J Rheumatol 1989; 18: 361-368.
-
Aabakken L, Larsen S, Osnes M. Cimetidine tablets or suspension for the prevention of gastrointestinal mucosal lesions caused by non-steroidal anti-inflammatory drugs. Scand J Rheumatol 1989; 18: 369-375.
-
Aabakken L, Bjørnbeth BA, Weberg R, Vikmoen L, Larsen S, Osnes M. NSAID-associated gastroduodenal damage does famotidine protection extend into the mid and distal duodenum ? Aliment Pharmacol Therap 1990; 4: 295-303.
-
Bjarnason I, Williams P, Smethurst P, Peters TJ, Levi AJ. Effect of non-steroidal anti-inflammatory drugs and prostaglandins on the permeability of the human small intestine. Gut 1986; 27: 1292-1297.
-
Bjarnason I, Smethurst P, Fenn CG, Lee CE, Menzies IS, Levi J. Misoprostol reduces indomethacin- induced changes in human small intestinal permeability. Dig Dis Sci 1989; 34: 407-411.
-
Bjarnason I. Experimental evidence of the benefit of misoprostol beyond the stomach in humans. J Rheumatol 1990; (Suppl 20) 17: 38-41.
-
Bjarnason I, Smethurst P, Clark P, Menzies I, Levi J, Peters T. Effect of prostaglandin on indomethacin-induced increased intestinal permeability in man. Scand J Gastroenterol Suppl 1989; 164: 97-102.
-
Davies GR, Wilkie ME, Rampton DS. Effects of metronidazole and misoprostol on indomethacin-induced changes in intestinal permeability. Dig. Dis. Sci. 1993; 38: 417-425.
-
Jenkins RT, Rooney PJ, Hunt RH. Increased bowel permeability to [51Cr]EDTA in controls caused by Naproxen is not prevented by cytoprotection. Arth Rheum 1988; 31 (Suppl 1): R11.
-
Morris AJ, Murray L, Sturrock RD, Madhok R, Capell HA, Mackenzie JF. Short report: the effect of misoprostol on the anemia of NSAID enteropathy. Aliment Pharmacol Ther 1994; 8: 343-346.
-
Bjarnason I, Hayllar J, Smethurst P, Price A, Gumpel MJ. Metronidazole reduces intestinal inflammation and blood loss in non-steroidal anti-inflammatory drug induced enteropathy. Gut 1992; 33: 1204-1208.
-
Müller M. Reductive activation of nitroimidazoles in anaerobic microorganisms. Biochem Pharmcol 1986; 35: 37-41.
-
Akamatsu H, Oguchi M, Nishijima S, Asada Y, Takahashi M, Ushijima T, Niwa Y. The inhibition of free radical generation by human neutrophils through the synergistic effects of metronidazole with palmitoleic acid: a possible mechanism of action of metronidazole in rosacea and acne. Arch Dermatol Res 1991; 282: 449-454.
-
Arndt H, Paltisch K-D, Grisham MB, Granger DN. Metronidazole inhibits leukocyte-endothelial cell adhesion in rat mesenteric venules. Gastroenterology 1994; 106: 1271-6.
-
Banerjee AK, Sherwood R, Rennie JA, Peters TJ. Sulphasalazine reduced indomethacin induced changes in small intestinal permeability in man. Brit Soc Gastro 1986; 23: A593.
-
Bjarnason I, Hopkinsson N, Zanelli G, Prouse P, Smethurst P, Gumpel JM, Levi AJ. Treatment of non-steroidal anti-inflammatory drug induced enteropathy. Gut 1990; 33: 777-780.
-
Hayllar J, Smith T, Macpherson A, Price AB, Gumpel M, Bjarnason I. Nonsteroidal antiinflammatory drug-induced small intestinal inflammation and blood loss. Effects of sulfasalazine and other disease-modifying antirheumatic drugs. Arthritis Rheum 1994; 37: 1146-50.
-
Cronstein BN, Naime D, Ostad E. The antiinflammaotyr mecahnism of methotrexate. Increased adenosine release at inflamed sites diminishes leukocyte accumulation in an in vivo model of inflammation. J Clin Invest 1993; 92: 2675-82
-
Pooley N, Ghosh L, Sharon P. Up-regulation of E-selectin an intercellular adhesion molecule-1 differs between Crohn’s disease and ulcerative colitis. Dig Dis Sci 1995; 40: 219-25.
-
Bjarnason I, Smethurst P, Macpherson A, Walker F, McElnay JC, Passmore P, Menzies IS. Glucose and citrate reduce the permeability changes caused by indomethacin in humans. Gastroenterology 1992; 102:1546-1550.
-
Sperling RI, Weinblatt M, Robin JL, Ravalese J 3rd, Hoover RL, House F, Coblyn JS, Fraser PA, Spur BW. Effects of dietary supplementation with marine fish oil on leukocyte lipid mediator generation and function in rheumatoid arthritis. Arthritis Rheum 1987; 30: 988-97.
-
Kremer JM, Malamood H, Maliakkal B, Rodgers JB, Ross JS, Cooper JA. Fish oil dietary supplementation for prevention of indomethacin induced gastric and small bowel toxicity in healthy volunteers. J Rheumatol 1996; 23: 1770-3.
-
Hond ED, Peeters M, Hiele M, Bulteel V, Ghoos Y, Rutgeerts P. Effect of glutamine on the intestinal permeability changes induced by indomethacin in humans. Aliment Pharmacol Ther 1999; 13: 679-85.
-
Masferrer JL, Zweifel BS, Manning PT, et al. Selective inhibition of inducible cyclooxygenase in vivo is antiinflammatory and nonulcerogenic. Proc Nat Acad Sci 1994; 91: 3228-32.
-
Gans KR, Galbraith W, Roman RJ, Haber SB, Kerr JS, Schmidt WK, Smith C, Hewes WE, Ackerman NR. Anti-inflammatory and safety profile of DuP697, a novel orally effective prostaglandin synthesis inhibitor. J Pharmacol Exper Ther 1990; 254: 180-7.
-
Seibert K, Zhang Y, Leahy K, Hauser S, Masferrer J, Perkins W, Lee L, Isakson P. Pharmacological and biochemical demonstration of the role of cyclooxygenase 2 in inflammation and pain. Proc Nat Acad Sci 1994; 91: 12013-17.
-
Riendeau D, Percival MD, Boyce S, et al. Biochemical and pharmacological profile of a tetrasubstituted furanone as a highly selective COX-2 inhibitor. Br J Pharmacol 1997; 121: 105-17.
-
Bjarnason I, Thodleifsson B. Gasterointestinal toxicity of non-steroidal anti-inflammatory drugs: the effect of nimesulide compared with naproxen on the human gastrointestinal tract. Rheumatology 1999; 39(Suppl. 1): 24-32.
-
Bjarnason I, Simon TJ, Sigthorsson G, Crane R, Hoover M, Bolognese J, Quan J. Cox-2 specific inhibtion with MK-0966 25 or 50 mg Q.D. does not increase intestinal permeability: a controlled study with placebo (PBO) and indomethacvin 50 mg T.I.D. J Am Gastroenterol 1998; 93: 1670 (Abstract 246).
-
Davies NM, Skjodt NM. Clinical pharmacokinetics of meloxicam. A cyclo-oxygenase-2 preferential nonsteroidal anti-inflammatory drug. Clin Pharmacokinet 1999; 36: 115-26
-
Hawkey C, Kahan A, Steinbruck K, et al. Gastrointestinal tolerability of meloxicam compared to diclofenac in osteoarthritis patients. International MELISSA Study Group. Meloxicam Large-scale International Study Safety Assessment. Br J Rheumatol 1998; 37: 937-45.
-
Dequeker J, Hawkey C, Kahan A, Steinbruck K, Alegre C, Baumelou E, et al. Improvement in gastrointestinal tolerability of the selective cyclooxygenase (COX)-2 inhibitor, meloxicam, compared with piroxicam: results of the Safety and Efficacy Large-scale Evaluation of COX- inhibiting Therapies (SELECT) trial in osteoarthritis. Br J Rheumatol 1998; 37: 946-51.
-
Davies NM, Wright MR, Jamali F. Antiinflammatory drug-induced small intestinal permeability: the rat is a suitable model. Pharm Res 1994; 11: 1652-6.
-
Ford J, Houston JB. Concentration-response relationships for three nonsteroidal anti-inflammatory drugs in the rat intestine. Hum Exp Toxicol 1995; 14: 573-9.
-
Ford J, Martin SW, Houston JB. Assessment of intestinal permeability changes induced by nonsteroidal anti-inflammatory drugs in the rat. J Pharmacol Toxicol Methods 1995; 34: 9-16.
-
Sigthorsson G, Jacob M, Wrigglesworth J, Somasundaram S, Tavares I, Foster R, Roseth A, Rafi S, Mahmud T, Simpson R, Bjarnason I. Comparison of indomethacin and nimesulide, a selective cyclooxygenase-2 inhibitor, on key pathophysiologic steps in the pathogenesis of nonsteroidal anti-inflammatory drug enteropathy in the rat. Scand J Gastroenterol 1998; 33: 728-35
-
Smecuol E, Sugai E, Maurino E, Vazquez S, Niveloni S, Pedreira S, Meddings J, Bai JC. Gastrointestinal permeability to non-steroid anti-inflammatory drugs. A prospective study comparing meloxicam to slow release indomethacin. Preliminary results. Gastroenterology 1998; 114: A1087.
-
Mohammed S, Croom DW 2nd. Gastropathy due to celecoxib, a cyclooxygenase-2 inhibitor. N Engl J Med 1999; 340: 2005-6.
-
Reuben SS, Steinberg R. Gastric perforation associated with the use of celecoxib. Anesthesiology 1999 ;91:1548-9
-
Conforti A, Donini M, Brocco G, De Soldato P, Benoni G, Cuzzolin L. Acute anti-inflammatory activity and gastrointestinal tolerability of diclofenac and nitrofenac. Agents and Actions 1993; 40(3-4): 176-80
-
Wallace JL, Muscara MN, McKnight W, Dicay M, Del Soldato P, Cirino G. In vivo antithrombotic effects of a nitric oxide-releasing aspirin derivative, NCX-4016. Thromb Res. 1999 Jan 1;93(1):43-50
-
Lichtenberger LM, Wang ZM, Romero JJ, Ulloa C, Perez JC, Giraud MN, Barreto JC. Non-steroidal anti-inflammatory drugs (NSAIDs) associate with zwitterionic phospholipids: insight into the mechanism and reversal of NSAID-induced gastrointestinal injury. Nat Med. 1995;1(2):154-8
-
Anand BS, Romero JJ, Sanduja SK, Lichtenberger LM Phospholipid association reduces the gastric mucosal toxicity of aspirin in human subjects. Am J Gastroenterol 1999 Jul;94(7):1818-22
-
Regtop HL, Biffin JR. Divalent metal complexes of indomethacin, compositions and medical methods of use thereof US Patent 5466824, 1995
- Weder JE, Hambley TW, Kennedy BJ, Lay PA, Maclachlan D, Bramley R, Delfs CD, Murray KS, Moubaraki B, Warwick B, Biffin JR, Regtop H. Anti-inflammatory dinulear coppe (II) complexes with indomethacin. Synthesis magnetism and EPR spectroscopy. Crystal structure of the NN, dimethylformamide adduct. Inorg Chem 1999; 38: 1736-44
Corresponding author: Dr. Neal M. Davies, Faculty of Pharmacy, University of Sydney, Sydney, New South Wales, Australia, 2006.
Published by the Canadian Society for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences.
http://www.ualberta.ca/~csps
CSPS Home | JPPS Home | Search | Subscribe to JPPS |