J Pharm Pharmaceut Sci (www.ualberta.ca/~csps) 3(2):234-258, 2000
Nano and Microparticles as Controlled Drug Delivery Devices
Manuscript received February 10th, 2000, Revised May 11th, 2000; Accepted June 26th, 2000.
Majeti N. V. Ravi
Kumar1
Department
of Chemistry, University
of Roorkee, India
PDF version for printing
ABSTRACT
Although, the drug delivery system (DDS) concept is not new, great progress has recently been made in the treatment of a variety of diseases. Targeting delivery of drugs to the diseased lesions is one of the most important aspects of DDS. To convey a sufficient dose of drug to the lesion, suitable carriers of drugs are needed. Nano and microparticle carriers have important potential applications for the administration of therapeutic molecules. The research in this area is being carried out all over the world at a great pace. Research areas cover novel properties that have been developed increased efficiency of drug delivery, improved release profiles and drug targeting. The purpose of this review is to take a closer look at nano/microparticles as drug delivery devices.
1. Introduction
Controlled drug delivery technology represents one of the frontier areas of science, which involves multidisciplinary scientific approach, contributing to human health care. These delivery systems offer numerous advantages compared to conventional dosage forms, which include improved efficacy, reduced toxicity, and improved patient compliance and convenience. Such systems often use macromolecules as carriers for the drugs. By doing so, treatments that would not otherwise be possible are now in conventional use. This field of pharmaceutical technology has grown and diversified rapidly in recent years. Understanding the derivation of the methods of controlled release and the range of new polymers can be a barrier to involvement from the nonspecialist. Of the different dosage forms reported, nanoparticles and microparticles attained much importance, due to a tendency to accumulate in inflamed areas of the body (1-3). Nano and microparticles for their attractive properties occupy unique position in drug delivery technology. Some of the current trends in this area will be discussed.
2. Nanoparticles and Nanospheres
Nanoparticles were first developed around 1970. They were initially devised as carriers for vaccines and anticancer drugs (4). In order to enhance tumor uptake, the strategy of drug targeting was employed, and as a first important step, research focused on the development of methods to reduce the uptake of the nanoparticles by the cells of the reticuloendothelial system (RES) (5). Simultaneously, the use of nanoparticles for ophthalmic and oral delivery was investigated (6).
2.1 Poly(ethylene oxide)-poly(L-lactic acid)/poly(b-benzyl-L-aspartate)
Polymeric micelles often self-assemble when block copolymers are used for their preparation (7). Micelles, based on the biocompatible copolymers of poly(ethylene oxide) PEO with poly(L-Lactic acid) PLA or with poly(b-benzyl-L-aspartate) PBLA, have been described in literature (8,9). Synthesis of such nanospheres with functional groups on their surface is shown in Figure 1.
Aldehyde groups on the surface of the PEO-PLA micelles may react with the lysine residues of cell’s proteins. They may also be used for attachment of the amino-containing ligands. The hydroxyl groups on the surface of the PEO-PBLA micelles can be further derivatized and conjugated with molecules capable to pilot the modified micelles to specific sites of living organism. Such nanospheres have been tested as vehicles for delivery of anti-inflammatory and anti-tumor drugs (10,11).
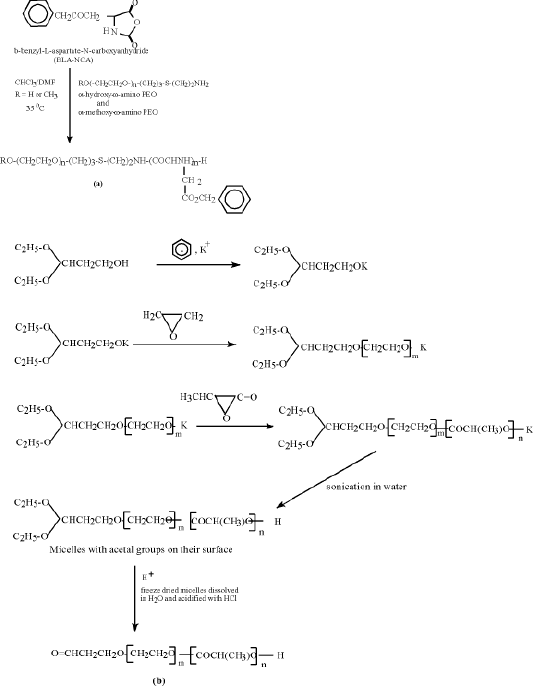
Figure 1 (a) poly(ethylene oxide)-co-b-benzyl-L-aspartate(PEO-PBLA) and (b) poly(ethylene oxide) -co-L-lactide (PEO-PLLa) micelles with aldehyde groups on their surface
2.2 Poly(lactide-co-glycolide)-[(propylene oxide)-poly(ethylene oxide)]
Nanoparticles (80-150 nm) of the biocompatible and biodegradable polyester copolymer PLG [Poly(lactide-co-glycolide)] Figure 2 have been reported (12,13) by the nanoprecipitation method (they have been precipitated with acetone from their oily colloidal nanodispersion in water). Thus formed particles of PLG were coated with 5-10 nm thick layer of the poly(propylene oxide) - poly(ethylene oxide) (PPO-PEO) block copolymer or with tetrafunctional (PEO-PPO)2 -N-CH2-CH2-N-(PPO-PEO)2 (12,13). Such coats are bound to the core of the nanosphere by the hydrophobic interactions of the PPO chains, while PEO chains protrude into the surrounding medium and form a steric barrier, which hinders the adsorption of certain plasma proteins onto the surface of such particles. On the other hand, the PEO coat enhances adsorption of certain other plasma compounds. In consequence, the PEO-coated nanospheres are not recognized by macrophages as foreign bodies and are not attacked by them (14).
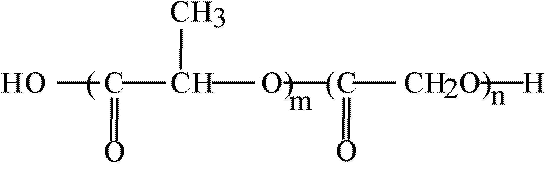
Figure 2 Poly(lactide-co-glycolide)PLG
2.3 Polyphosphazene derivatives
Allock and coworkers developed derivatives of the phosphazene polymers suitable for biomedical applications (15-17). Long-circulating in the blood, 100-120 nm in diameter, PEO-coated nanoparticles of the poly(organophospazenes) containing amino acid, have been prepared. PEO-polyphosphazene copolymer, or poloxamine 908 (a tetrafunctional PEO copolymer) has been deposited on their surface (18,19). Chemical formulae of such polyphosphazene derivatives are shown in Figure 3.
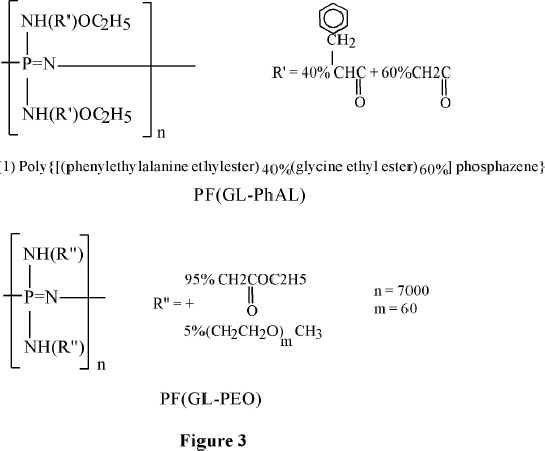
Figure 3 Polyphosphazenes for medical applications
2.4 Poly(ethylene glycol) coated nanospheres
Poly(ethylene glycol) PEG-coated nanospheres from PLA, PLG, or other biodegradable polymers viz., poly(e-caprolactone) (PCL), may be used for the intravenous drug delivery. PEG and PEO denote essentially identical polymers. The only difference between the respective notations is that methoxy groups in PEO may replace the terminal hydroxyls of PEG. It has been pointed out that PEG coating of nanospheres provides protection against interaction with the blood components, which induce removal of the foreign particles from the blood. It prolongs, therefore, their circulation in the blood stream. In consequence, thus coated nanospheres may function as circulation depots of the administered drugs (20,21). Slowly releasing drugs into plasma, and thus altering their concentration profiles can achieve obvious therapeutical benefits. About 200 nm in diameter PEG-coated nanospheres, in which PEG is chemically bound to the core have been prepared, in the presence of monomethoxy PEG, by ring opening polymerization (with stannous octoate as a catalyst) of such monomers as e-caprolactone, lactide, and/or glycolide (21). Ring opening polymerization of these monomers in the presence of such multifunctional hydroxy acids as citric or tartaric, to which several molecules of the monomethoxy monoamine of PEG (MPEG-NH2) have been attached, yields multiblock (PEG)n-(X)m copolymers. PEG-PLA copolymer in which NH2 terminated methoxy PEG molecules have been attached to tartaric acid is shown schematically in Figure 4.
It has been demonstrated that morphology, degradation, and drug encapsulation behavior of copolymers containing PEG blocks strongly depends on their chemical composition and structure. Studies of nanoparticles composed of the diblocks of the poly(DL-lactide-co-glycolide) with the methoxy terminated poly(ethylene glycol) [PLG-PEG] or of the branched multi-blocks PLA-(PEG)3, in which three methoxy terminated PEG chains are attached through a citric acid residue, suggested that they have a corecorona structure in an aqueous medium. The polyester blocks form the solid inner core. The nanoparticles, prepared using equimolar amounts of the PLLA-PEG and PDLA-PEG stereoisomers, are shaped as discs with PEG chains sticking out from their surface.
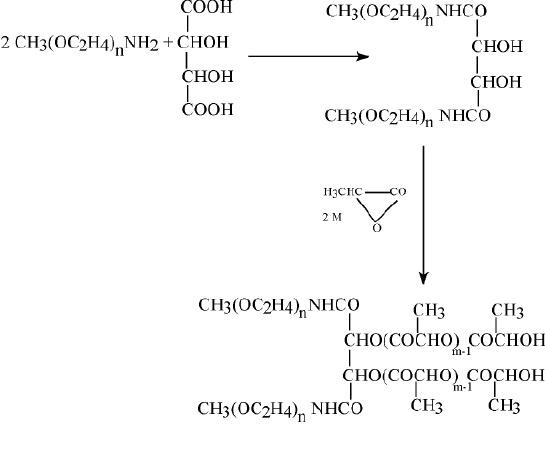
Figure 4 Multiblock (PEG)n-(x)m copolymers. Amino terminated methoxy polyethylene glycol molecules attached to tartaric acid with Pla side chains
Their hydrophobic/hydrophilic content seems to be just right for applications in cancer and gene therapies (7). Such nanospheres are prepared by dispersing the methylene chloride solution of the copolymer in water and allowing the solvent to evaporate (21).
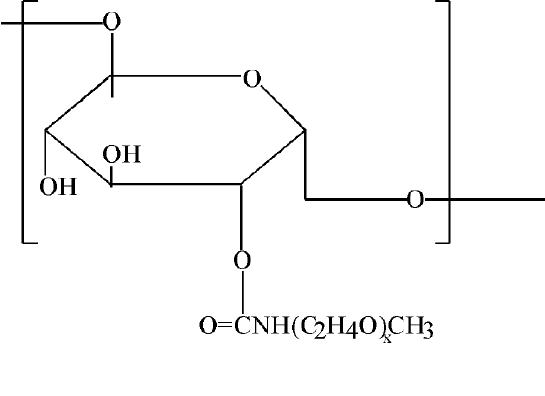
Figure 5 PEG-dextran conjugates
By attaching biotin to its free hydroxyl groups and complexing it with avidin, cell specific delivery may be attained. NMR studies of such systems (22) revealed that the flexibility and mobility of the thus attached PEG chains is similar to that of the unattached PEG molecules dissolved in water. Recently, PLG microspheres, with the PEG-dextran conjugates Figure 5, attached to their surface, have been investigated as another variant of the above-described approach. Microspheres of diameter 400-600 nm have been prepared (23). To the glycopyranose hydroxyls of the dextran units, targeting moieties can be attached (23).
2.5 Azidothymidin (AZT)/dideoxycytidine (ddc) nanoparticles
Recently, research took advantage of the properties of nanoparticles, to be easily taken up by cells of the RES (3). In the case of AIDS, for instance, the macrophages of the RES represent one of the most important therapeutic targets (24). In addition to the CD4+T lymphocyles, these cells play a decisive role as a reservoir for the human immunodeficiency virus (HIV). In tissues such as the lung and the brain, HIV is located primarily in macrophage-like cells (i.e., alveolar macrophages and microglia, respectively (25,26). In contrast to CD4+ T cells, in which HIV replication is proliferation dependent and finally leads to cell death, macrophages in a mature nonproliferating but immunologically active state can be productively infected with HIV type 1 (HIV-1) and HIV-2 (27-29). Altered cellular functions in the macrophage population may contribute to the development and clinical progression of AIDS. A large proportion of AIDS patients show HIV encephalitis with diffuse neuronal damage which is thought to be mediated by viral proteins and/or factors with neurotoxic activity (i.e. cytokines). This can occur through proinflammatory cytokines or by HIV-1-specific proteins secreted from cells of the mononuclear phagocyte system, including brain macrophages, microglia and multinucleated giant cells, which have been shown to be productively infected with HIV (30-32).
It is evident that, apart from having a function in the pathogenesis of the disease cells of the macrophage lineage are vectors for the transmission of HIV. The placental macrophage is likely to be the primary cell type responsible for vertical (maternofetal) transmission of HIV (33). For mucosal transmission it was found that an important property of the transmitted HIV variant is its ability to infect macrophages (34). The phenotypic characterization of virus populations which were transmitted sexually or vertically a selection of variants with a predominate tropism for macrophage in the recipient (35). Because of the important role of cells of the monocyte/macrophage (Mo/Mac) lineage in the pathogenesis of HIV, fully effective anti-HIV therapy must reach Mo/Mac in addition to other cells (32).
Consequently, nanoparticles without surfactant coating leading to stealth properties may represent promising drug carriers for this disease. Therefore, the potential of nanoparticles to deliver antiviral drugs to HIV-infected and uninfected macrophages was investigated by Schafer et al (36,37). It was found in in vitro cell cultures of human moncytes/macrophages (Mo/Mac) that the uptake of nanoparticles by HIV-1-infected cells was about 30% higher after 7 days of culture and about 65% after 21 days than by uninfected cells. Similar results were observed with macrophages obtained from AIDS patients, depending on the state of disease. The uptake by these cells was also dependent on the surface coating with different surfactants (37). Interestingly, despite their possible stealth properties, these surfactants did not reduce the in vitro uptake of the nanoparticle: ploxamer 338 had no influence and poloxamer 188 even increased the uptake (36,37).
The following investigation of the in vitro inhibition of the development of infection of the above human macrophage cultures by HIV-1 using the antiviral drugs azidothymidin (AZT) and dideoxycytidine (ddc), however, revealed no difference between free and nanoparticle-bound drugs (38): nanoparticle-bound AZT or ddc retained their activity but were not superior to free drug. The situation was totally different with the antiviral protease inhibitor saquinavir (32). Here a 10-fold increase in efficacy of nanoparticle-bound drug over free drug was observed. The difference in these results probably is due to the fact that AZT and ddc easily penetrate into cell uptake by the nanoparticles.
However, it has to be considered that in tissue cultures a homogenous cell population exists, which is totally different to the situation in the human or animal body: the body consists of a huge number of different cell types. Macrophages represent only a small percentage of the total number of cells. As a consequence, it was conceivable that even with AZT or ddc nanoparticles still may achieve a better targeting to these cells despite their inability to show a better antiviral efficacy of these drugs in vitro in the homologous cell cultures. Indeed that was observable: Lobenberg and Kreuter (39) bound 14C-labeled AZT to nanoparticles. After i.v. injection of nanoparticle-bound drug, the concentration of the AZT-label in the organs that were rich in macrophages, such as the liver (39), were up to 18 times higher than with AZT solution. Likewise, after oral administration the nanoparticle formulation delivered the AZT-label more efficiently to the RES than the aqueous solution. In addition, the blood concentration was significantly higher after oral administration of nanoparticles.
Autoradiographic investigations using radioluminography supported the above results (40). The radioactivity in the liver, lung, and spleen, organs with a high number of macrophages was much higher than in other parts of the body with the nanoparticles formulation. The above organs showed a spotted appearance of the radioactivity that is typical for accumulation in macrophages after i.v. as well as after oral administration of the drug bound to the particles. In contrast, after administration of the solution, the radioactivity in the above organs was homogeneously distributed and much lower. These results show that the nanoparticles seemed to have reached their target and may represent very promising delivery system for AIDS therapy.
2.6 Poly (isobutylcynoacrylate) nanocapsules
Intragastric administration of insulin-loaded poly(isobutylcyanoacrylate) nanocapsules induced a reduction of the glycemia to normal level in streptozotocin diabetic rats (41-43) and is alloxan induced diabetic dogs (44). The hypolglycemic effect was characterized by surprising events including a lag time period of 2 days and a prolonged effect over 20 days. Insulin is a very hydrosoluble peptide and should be inactivated by the enzymes of the gastrointestinal tract. Thus, the reason why insulin could be encapsulated with high efficiency in nanocapsules containing an oily core and why these nanocapsules showed so unexpected biological effect remained unexplained. Nanocapsules were prepared by interfacial polymerization of isobutylcyanoacrylate (45). Any nucleophilic group including those of some of the aminoacids of insulin (46) could initiate the polymerization of such a monomer. In this case insulin could be found covalently attached to the polymer forming the nanocapsule wall as it was recently demonstrated with insulin-loaded nanosphers (47).
Aboubakar et al., (48) studied, physico-chemical characterization of insulin-loaded poly(isobutyl cyanoacrylate) nanocapsules obtained by interfacial polymerization. They claimed that the large amount of ethanol used in the preparation of the nanocapsules that initiated the polymerization of isobutylcyanoacrylate preserving the peptide from a reaction with monomer resulting high encapsulation rate of insulin. From their investigations, it appears that insulin was located inside the core of the nanocapsules and not simply adsorbed onto their surface.
2.7 Poly(g-benzyl-L-glutamate)/poly(ethylene oxide)
Nanoparticles have been widely investigated as the drug carriers (49-52). Biodegradable poly(D,L-lactide) (53,54) polybutylcyanoacrylate (55) and poly(e-caprolactone) (56) are widely being used to prepare nanoparticles. The advantages of the nanoparticles are the reduced drug toxicity, the improvement of biodistribution, and the increased therapeutic efficacy. Diblock copolymers have been studied in the sustained release system as an alternative drug carrier (57,58), since they are known to form a micelle structure. Hydrophilic-hydrophobic diblock copolymers exhibit amphiphilic behavior and form micelles with core-shell architecture. These polymeric carriers have been used to solubilize hydrophobic drugs, to increase blood circulation time, to obtain favorable biodistribution and to lower interactions with reticuloendothelial system (59-61). In the same direction, Oh et al (62) reported the preparation and characterization of polymeric nanoparticles containing adriamycin as a model drug. The nanoparticles are obtained from poly(g-benzyl-L-glutamate)/poly(ethylene oxide) [PBLG/PEO] diblock copolymer, which form a hydrophobic inner core and a hydrophilic outer shell of micellar structure (63,64), by adopting dialysis procedure. Their results indicate that only 20% of the entrapped drug was released in 24 h at 37 0C and the release were dependent on the molecular weight of hydrophobic polymer.
2.8 Chitosan-poly(ethylene oxide) nanoparticles
Hydrophilic nanoparticle carriers have important potential applications for the administration of therapeutic molecules (7,24). Most of the recently developed hydrophobic-hydrophilic carriers require the use of organic solvents for their preparation and have a limited protein-loading capacity (19,65-67). Calvo et al (68) reported a new approach for the preparation of nanoparticles, made solely of hydrophilic polymer, to address these limitations. The preparation technique, based on an ionic gelation process, is extremely mild and involves the mixture of two aqueous phases at room temperature.
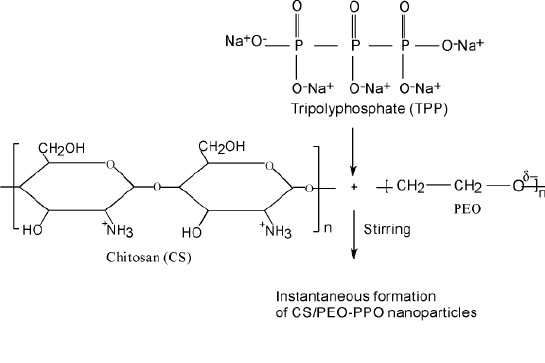
Figure 6 The preparation of CS nanoparticles- A schematic diagram
One phase contains the polysaccharide chitosan (CS) and a diblock copolymer of ethylene oxide and polyanion sodium tripolyphosphate (TPP) Figure 6. It was stated that, the size (200-1000 n) and zeta potential (between + 20mv and +60mv) of nanoparticles can be conventionally modulated by varying the ratio CS/PEO-PPO. Furthermore, using bovine serum albumin (BSA) as a model protein, it was shown that these new nanoparticles have great protein loading capacity (entrapment efficiency up to 80% of the protein) and provide a continuous release of the entrapped protein for up to 1 week (68).
2.9 Methotrexate-o-carboxymethylate chitosan
Nanoparticles of methotrexate (MTX) were prepared using o-carboxymethylate chitosan (o-CMC) as wall forming materials, and an isoelectric-critical technique under ambient condition (64). Drug controlled releases were studied in several media including simulated gastric fluid, intestinal fluid and 1% fresh mice serum. It was found that acidic media provide a fast release rate than neutral media. The effect of MTX/o-CMC ratio and amount of crosslinking agents of drug release in different media were evaluated. The changes of size and effective diameter of o-CMC nanoparticles were detected by SEM and laser light scattering system before and after the drug release. The author claimed that, the o-CMC nanoparticles constitute an attractive alternative to other anticancer drugs and enzyme carriers (69).
2.10 Solid lipid nanoparticles (SLNs)
Solif lipid nanoparticles (SLNs), one of the colloidal carrier systems, has many advantages such as good biocompatibility, low toxicity and stability (70). Schwarz and Mehnert (71) studied the lipophilic model drugs tetracaine and etomidate. The study highlights the maximum drug loading, entrapment efficiacy, effect of drug incorporation on SLN size, zeta potential (charge) and long-term physical stability. Drug loads of up to 10% were achieved, while simultaneously maintaining a physical stable nanoparticle dispersion (71). They claimed that the incorporation of drugs showed no or little effect on particle size and zeta potential compared to drug free SLN (71). In another study, Kim and Kim (72) studied the effect of drug lipophilicity and surfactant on the drug loading capacity, particle size and drug release profile. The prepared SLNs by homogenization of melted lipid dispersed in an aqueous surfactant solution. Ketoprofen, ibuprofen and pranoprofen were used as model drugs and tween and poloxamer surfactants were tested (72). Mean particle size of prepared SLNs was ranged from 100 to 150 nm. It was found that the drug loading capacity was improved with the most lipophilic drug and low concentration of surfactant (72).
3. Microcapsules and microSpheres
The term “microcapsule” is defined, as a spherical particle with the size varying in between 50 nm to 2 mm containing a core substance. Microspheres are in strict sense, spherically empty particles. However, the terms microcapsules and microspheres are often used synonymously. In addition, some related terms are used as well. For example, “microbeads” and “beads” are used alternatively. Sphere and spherical particles are also employed for a large size and rigid morphology. Due to attractive properties and wider applications of microcapsules and microspheres, a survey of the applications in controlled drug release formulations is appropriate.
3.1 Multiporous beads of chitosan
Several researchers (73,74) have studied simple coacervation of chitosan in the production of chitosan beads. In general, chitosan is dissolved in aqueous acetic acid or formic acid. Using a compressed air nozzle, this solution is blown into NaOH, NaOH-methanol, or ethanediamine solution to form coacervate drops. The drops are then filtered and washed wit hot and cold water successively. Varying the exclusion rate of the chitosan solution or the nozzle diameter can control the diameter of the droplets. The porosity and strength of the beads correspond to the concentration of the chitosan-acid solution, the degree of N-deacetylation of chitosan, and the type and concentration of coacervation agents used.
The chitosan beads described above have been applied in various fields viz., enzymatic immobilization, chromatograph support, adsorbent of metal ions, or lipoprotein, and cell cultures. It was confirmed that the porous surfaces of chitosan beads make good cell culture carrier. Hayashi and Ikada (75), immobilized protease onto the porous chitosan beads which carry active groups with a spacer and found the immobilized protease had higher pH, thermal storage stability, and gave rather higher activity toward the small ester substrate, N-benzyl-L-arginine ethyl ester. In addition, Nishimura et al (73) investigated the possibilities of using chitosan beads as a cancer chemotherapeutic carrier for adriamycin. Recently, Sharma et al (76-78) prepared chitosan microbeads for oral sustained delivery of nefedipine, ampicillin and various steroids by adding to chitosan and then going through a simple coacervation process. These coacervate beads can be hardened by crosslinking with glutaraldehyde or epoxychloropropane to produce microcapsules containing rotundine (79). The release profiles of the drugs from all these chitosan delivery systems were monitored and showed in general the higher release rates at pH 1-2 than that at pH 7.2-7.4. The effect of the amount of drug loaded, the molecular weight of chitosan and the crosslinking agent on the drug delivery profiles have been discussed (76-79).
3.2 Coated alginate microspheres
Many of the present controlled release devices for in vivo delivery of macromolecular drugs involve elaborate preparations, often employing either harsh chemicals, such as organic solvents or extreme conditions, such as elevated temperature. The conditions have the potential to destroy the activity of sensitive macromolecular drugs, such as proteins or polypeptides. In addition, many devices require surgical implantation and in some cases, the matrix remains behind or must be surgically removed after the drug is exhausted (80).
Wheatley et al (81) studied a mild alginate/polycation microencapsulation process, as applied to encapsulation of bioactive macromolecules such as proteins. The protein drugs were suspended in sodium alginate solution and sprayed into 1.3% buffered calcium chloride to form cross-linked microcapsules, large (upto 90%) losses of encapsulation species were encountered, and moderate to strong protein-alginate interactions caused poor formation of capsules. As a result, a diffusion-filling technique calcium alginate microcapsules were formed by spraying 10 ml of the sodium alginate solution into 250 ml of buffered 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethane sulfonic acid (HEPES) calcium chloride (13 MM HEPES, 1.3% CaCl2 pH 7.4) from a 20 ml plastipack syringe through a 22 G needle Figure 7.
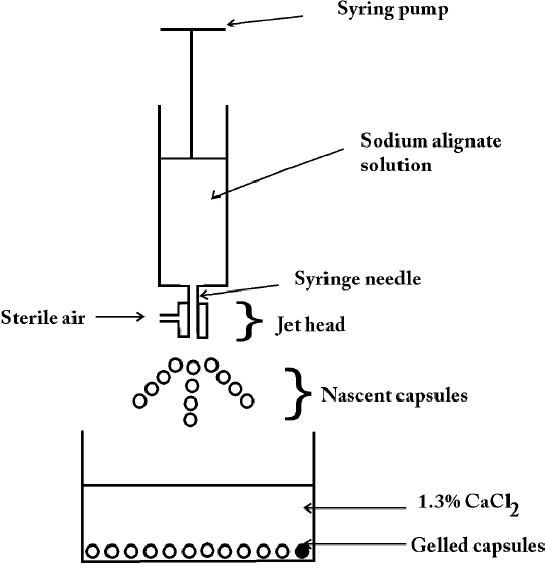
Figure 7 Schematic of spray device used in the preparation of calcium alginate microsphere
Protein was then loaded into these capsules by stepwise diffusion from solutions of increasing drug concentration. The drug loaded capsules were coated with a final layer of polycation. In all, three polycation coatings were used, two prior filling and one after filling. The first coating strongly influenced the size, integrity, and loading capacity of the capsules. Low concentrations of polycation resulted in poorly formed capsules with very low retention of the drug in the final capsule, while very high concentration prevented the drug from entering the capsule at the filling stage. The first coat also affected the duration of drug release from the capsule and the size of the burst effect. The second coat had less effect on the capsule integrity, but it did influence the drug payload and release profile. The final, sealing-coat had little effect on drug payload and only limited effect on the release profile upto a critical concentration, above which the release profile was not affected. For all coats, increasing polycation concentration decreased the burst effect, and caused the release profile to be more sustained. Encapsulation of a series of dextrans with increasing molecular weight revealed that the release profile was directly related to the molecular weight of the diffusing species, which was more sustained as molecular weight increased. More recently, Murata et al (82) investigated alginate gel bead containing chitosan salt. When the bead was placed in bile acid solution it rapidly took bile acid into itself. The uptake amount of taurocholate was about 25 m mol/0.2 g dried gel beads. The phenomenon was observed on the case of the beads incorporating colestyramine instead of chitosan. From the studies reported, it appears that ion-exchange reaction accompanying the insoluble complex-formation between chitosan salt and bile acid occurs in alginate gel matrix (82).
3.3 N-(aminoalkyl) chitosan microspheres
The most promising encapsulation system yet developed appears to be the encapsulation of calcium alginate beads with poly-L-lysine. However, the use of this system on a large scale, such as for oral vaccination of animals, is not feasible due to the high cost ($200/g) of poly-L-lysine (PLL). It would therefore be desirable to develop an economic and reliable microencapsulation system based on chitosan and alginate. The better membrane-forming properties of PLL over chitosan were for the following reasons: PLL contains a number of long-chain alkylamino groups that extend from the polyamide backbone. These chains may extend in a number of directions and interact with various alginate molecules, through electrostatic interactions, resulting in a highly crosslinked membrane. Chitosan, on the other hand, has aminogroups that are very close to the polysaccharide backbone. Interaction between the charged amino groups of chitosan and carboxylate groups of alginate may be lessened due to steric repulsion between the two molecules.
Goosen and coworkers (83) attempted to mimic the properties of PPL by extending the length of the cationic spacer arm on the chitosan main chain. In the chemical modification, chitosan was first reacted with a-bromoacylbromide followed by reaction with an amine. The major problem in this procedure was the competing hydrolysis reaction of the bromoacylbromide. Furthermore, the lack of characterization of the modified chitosan caused ambiguity in the effectiveness of the chitosan modification. No significant difference was found in membrane properties between modified and unmodified chitosan. A two-step synthetic method for attaching long alkylamine side chains to chitosan is represented in Figure 8.
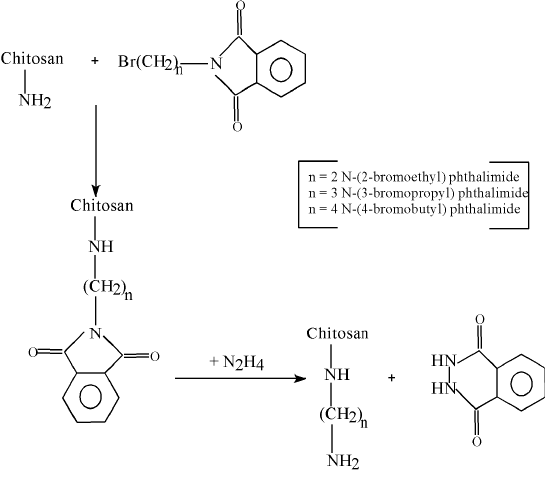
Figure 8 Modification of chitosan with bromoalkylphthalimides and hydrazine
The approach outlined in Figure 8 is designed to attach flexible alkylamine side chains to the chitosan polysaccharide backbone, possibly simulating the behavior of PPL. The presence of two amino groups in this side chain may even enhance membrane-forming properties. Chemical modification of poly(vinyl alcohol) (PVA) by a similar procedure may also produce a- polyamine with membrane-forming properties similar to that of PPL (83). All alkylation products were characterized by solution 1H- and 13C-NMR and by solid-state CP-MAS 13C-NMR. The above synthetic polymer derivatives, as well as chitosan, polyallylamine, and polyethyleneimine, were used to form membrane coatings around the calcium alginate beads in which blue dextran of molecular weight 7.08x104 or 26.6x104 was entrapped. These microcapsules were prepared by extrusion of a solution of blue dextran in sodium alginate into a solution containing calcuim chloride and the membrane polymer. Measuring the elution of the blue dextran from the capsules, spectrophotometrically (83), assessed membrane integrity and permeability.
3.4 Chitosan/calcium alginate beads
The encapsulation process of chitosan and calcium alginate as applied to encapsulation of hemoglobin was reported by Huguet et al [84]. In the first process, the mixture of hemoglobin and sodium alginate is added dropwise to the solution of chitosan and the interior of capsules thus formed in the presence of CaCl2 is hardened. In the second method, the droplets were directly pulled off in a chitosan- CaCl2 mixture. Both procedures lead to beads containing a high concentration of hemoglobin (more than 90% of the initial concentration (150 g/L) were retained inside the beads) provided chitosan concentration is sufficient.
The molecular weight of chitosan (mol wt 245000 or 390000) and the pH (2, 4, or 5.4) had only a slight effect on entrapment of hemoglobin, the best retention being obtained with beads prepared at pH 5.4. The release of hemoglobin during the bead storage in water was found to be dependent on the molecular weight of chitosan. The best retention during storage in water was obtained with beads prepared with the high molecular weight chitosan solution at pH 2.0. Considering the total loss in hemoglobin during the bead formation and after 1 month of storage in water, the best results were obtained by preparing the beads in an 8 g/L solution of a 390000 chitosan at pH 4 (less than 7% of loss with regard to the 150 g/L initial concentration).
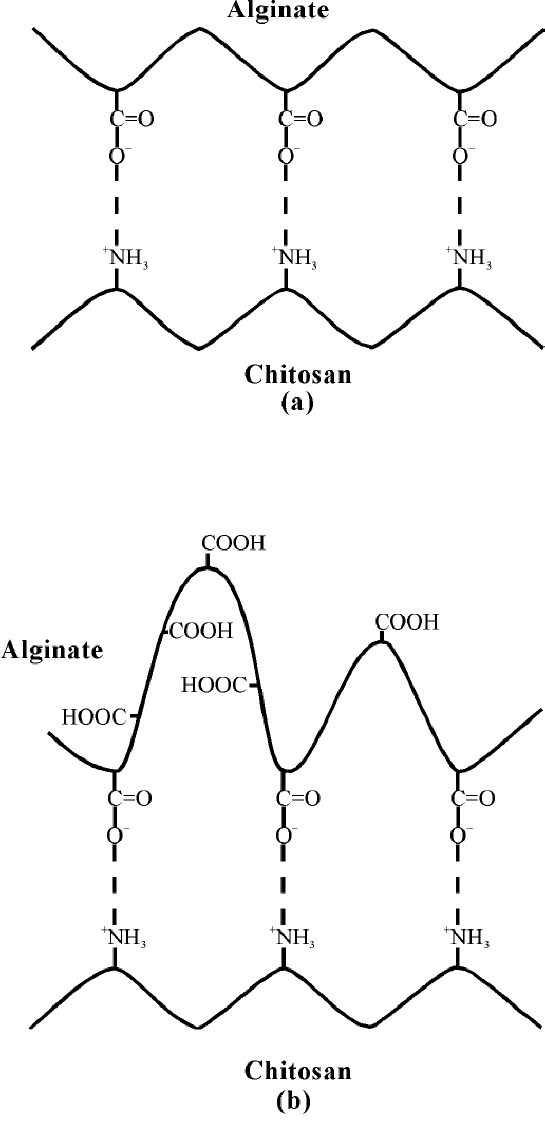
Figure 9 Schematic representation of the ionic interactions between alginate and chitosan (a) pH 5.4; (b) pH 2.0
Similarly, the encapsulation of various molecules [Hb, bovine serum albumin (BSA) and dextrans with various molecular weights] in calcium alginate beads coated with chitosan has been reported [85,86]. Their release has been compared and the influence of the conformation, the chemical composition and the molecular weight of the encapsulated materials have been analysed [85]. The ionic interaction between alginate and chitosan at different pH are depicted in Figure 9.
3.5 Poly(adipic anhydride) microspheres
In ocular drug delivery, the high rate of tear turnover, and the blinking action of the eyelids lead to short precorneal residence times for applied eye drops. Typically, the washout rate reduces the concentration of the drug in a tear film to one-tenth of its starting value in 4-20 min. As a result, only the eye absorbs a few percent of the administered drug and the duration of the therapeutic action may be quite short. Early reports showed that the formulation of the eye drops is decisive for the availability of an ocular drug (87). The absorbed amount of the model substance, fluoro metholone, and duration in the aqueous humor increased when a suspension was used instead of a solution, and the best result was obtained when the drug was formulated in an ointment. Similar results as with an ointment were obtained when a suspension formulated in a hydrogel was used (88). The combination of particles with a hydrogel thus increases the bioavailability of an ocular drug. A hydrogel may increase the precorneal residence time of a suspension, and the residence time of the hydrogel, therefore, sets the maximal achievable residence time for a given drug. Water-soluble drugs are generally not retained by hydrogels because of their high diffusion coefficients. One way of solving this problem is to incorporate these drugs into polymeric microparticles.
A novel microsphere-gel formulation was investigated aiming to extend precorneal residence times for ocular drugs (89). Poly(adipic anhydride) was used for microencapsulation of timolol maleate. A nonaqueous method for the microsphere preparation was employed due to the hydrolytical sensitivity of the polymer. Microspheres were prepared with an average diameter of 40 mm. The polymer and the microspheres were characterized before and during degradation using size exclusion chromatography (SEC), differential scanning calorimeter (DSC), X-ray diffraction, infrared spectroscopy (IR), and scanning electron microscopy (SEM) (89). The microspheres had a smooth external surface and a hallow center surrounded by a dense outer shell. Degradation of the microspheres resulted in a constant release of adipic acid, the degradation product, indicating a surface-eroding degradation mechanism. The surface erosion of the polymer controlled the release of incorporated substance, timolol maleate. The drug release rate profile appeared to be suitable for ocular drug delivery. However, the initial drug release rate was decreased to some extent when the PAA-microspheres were incorporated into an in situ gelling polysaccharide (89), GelriteÒ. The authors claimed that the improved ocular bioavailability of these novel microsphere-gel delivery formulations remains to be compared with that of ordinary eye drops.
3.6 Gellan-gum beads
Gellan gum is a linear anionic polysaccharide produced by the microorganism pseudomonas elodea (90,91). The natural form of the polysaccharide consists of a linear structure repeating tetrasaccharide unit of glucose, glucuronic acid and rhamnose (91-93) in a molar ratio of 2:1:1. Native gellan is partially acylated with acetyl and L-glyceryl groups located on the same glucose residue (94). X-ray diffraction analysis shows that gellan gum exists as a half-staggered, parallel, double helix which is stabilized by hydrogen bonds involving the hydroxymethyl groups of one chain and both carboxylate and glyceryl groups of the other (95). The presence of acetyl or glyceryl groups does not interfere with double formation but does alter its ion-binding ability. The commercial gellan gum is the deacetylated compound obtained by treatment with alkali (90), yielding the gum in its low acyl form in which the acetate groups do not interfere with helix aggregation during gel formation. Gellan forms gels in the presence of mono and divalent ions, although its affinity for divalent ions is much stronger (96). Milas et al (97), showed a mechanism of gelation based on aggregation of the double helix controlled by the thermodynamics of the solution in which the nature of the counter-ion is of prime importance. The apparent viscosity of the gellan gum dispersions can be markedly increased by increases in both pH and cation concentration (98,99). Gellen gum is mainly used as a stabilizer or thickening agent and it has a wide variety of applications, particularly in food industry (100,101), as a bacterial growth media (102,103) and in plant tissue culture (104). Its medical and pharmaceutical uses are in the field of sustained release. Due to its characteristic property of temperature-dependent and cation-induced gelation, gellan has been used in the formulation of eye drops, which gelify on interaction with the sodium ions naturally present in the eye fluids (104-107).
Microcapsules containing oil and other core materials have been formed by complex coacervation of gellan gum-gelatin mixture (108). Deacetylated gellan gum was used to produce a bead formulation containing sulphamethizole by a hot extrusion process into chilled ethylacetate (100). Recently, the ability of gellan gum to form gels in the presence of calcium ions enabled capsules to be prepared by gelation of the polysaccharide around a core containing starch (109-111), or oil was investigated.
More recently, Kedzierewicz et al (112) adopted a method rather simpler than the ones used so far, i.e. the ionotropic gelation method, to prepare gellan gum beads. Gellan gum beads of propranolol-hydrochloride, a hydrophilic model drug were prepared by solubilizing the drug in a dispersion of gellan gum and then dropping the dispersion into calcium chloride solution. Major formulation and process variables, which might influence the preparation of the beads and drug release from gellan gum beads were studied. Very high entrapment efficiencies were obtained (92%) after modifying the pH of both the gellan gum dispersion and the calcium chloride solution. The beads could be stored for 3 weeks in a wet or dried state without modification of the drug release. Oven-dried beads released the drug somewhat more slowly than the wet or freeze-dried beads. The drug release from the oven-dried beads was slightly affected by the pH of the dissolution medium (112). Gellan gum could be a useful carrier for the encapsulation of fragile drugs and provide new opportunities in the field of bioencapsulation.
3.7 Poly(D, L-lactide-co-glycolide) microspheres
The treatment of infiltrating brain tumors, particularly oligodendrogliomas, requires radiotherapy, which provides a median survival of 3.5-11 years (113). Since 5-iodo-21-deoxyuridine (IdUrd) is a powerful radio sensitizer (114), the intracarnial implantation of IdUrd loaded microparticles within the tumor might increase the lethal effects of g-radiations of malignant cells having incorporated IdUrd. The particles can be administered by stereotactic injection, a precise surgical injection technique (115). This approach requires microparticles of 40-50 mm in size releasing in vivo their content over 6 weeks, the standard period during which a radiotherapy course must be applied.
The solvent evaporation process is commonly used to encapsulate drugs into poly(lactide-co-glycolide) microparticles (PLGA) (116). It is well known that the candidate drugs must be soluble in the organic phase. In the case, where the active ingredient is not oil soluble, other alternative can be considered. The W/O/W-multiple emulsion method is particularly suitable for the encapsulation of highly hydrophilic drugs. For drugs which are slightly water soluble, like IdUrd (2mg/ml), other approaches must be investigated to achieve significant encapsulation: dissolution of the drug in the organic phase through the use of a cosolvent or dispersion of drug crystals in the dispersed phase. In the latter case, it is often admitted that the suspension of crystals in the organic phase can lead to an initial drug release, which is difficult control (117,118). To reduce IdUrd particle size, two-grinding processes were used, spray-drying and planetary ball milling (119-121). The optimal conditions of grinding were studied through experimental design and the impact on in vitro drug release from PLGA microspheres was then examined. More recently, Geze et al (122), studied IdUrd loaded poly(D,L-lactide-co-glycolide) (PLGA) microspheres with a reduced initial burst in the in vitro release profile, by modifying the drug grinding conditions. IdUrd particle size reduction has been performed using spray drying or ball milling. Spray drying significantly reduced drug particle size with a change of the initial crystalline form to an amorphous one and lead to a high initial burst. Conversely, ball milling did not affect the initial Id Urd crystallinity. Therefore, the grinding process was optimized to emphasize the initial burst reduction. The first step was to set qualitative parameters such as ball number, and cooling with liquid nitrogen to obtain a mean size reduction and a narrow distribution. In the second step, three parameters including milling speed, drug amount and time were studied by a response surface analysis. The interrelationship between drug amount and milling speed was the most significant factor. To reduce particle size, moderate speed associated with a sufficient amount of drug (400-500 mg) was used. Id Urd release from microparticles prepared by the O/W emulsion/extraction solvent evaporation process with the lowest crystalline particle size (15.3 mm) was studied to overcome burst effect. In the first phase of drug release, the burst was 8.7% for 15.3 mm compared to 19% for 19.5 mm milled drug particles (122).
In the other procedure, Rojas et al (123); optimized the encapsulation of b-lactoglobulin (BLG) within PLGA microcaparticles prepared by the multiple emulsion solvent evaporation method. The role of the pH of the external phase and the introduction of the surfactant Tween 20, in the modulation of the entrapment and release of BLG from microparticles, was studied. Better encapsulation of BLG was noticed on decreasing the pH of external phase to a value close to the PI of BLG, however, a larger burst release effect. In contrast, the addition of Tween 20 increased the encapsulation efficiency of BLG and considerably reduces in the burst release effect. In addition, Tween 20 reduced the number of aqueous channels between the internal aqueous droplets as well as those communicating with the external medium. Inventors claimed that these results constitute a step ahead in the improvement of an existing technology in controlling protein encapsulation and delivery from microspheres prepared by the multiple solvent evaporation method (123).
Blanco-Prieto et al (124) studied the in vitro release kinetics of peptides from PLGA microspheres, optimizing the test conditions for a given formulation, which is customary to determine in vitro/in vivo correlation. The somatostatin analogue vapreotide pamoate, an octapeptide, was microencapsulated into PLGA 50:50 by spray drying. The solubility of this peptide and its in vitro release kinetics from the microspheres were studied in various test media. The solubility of vapreotide pamoate was approximately 20-40 mg/ml in 67 mM phosphate buffer saline (PBS) at pH 7.4, but increased to 500-1000 mg/ml at a pH of 3.5. At low pH, the solubility increased with the buffer concentration (1-66 mM). Very importantly, proteins (aqueous bovine serum albumin (BSA) solution or human serum) appeared to solubilize the peptide pamoate, resulting in solubilities ranging from 900 to 6100 mg/ml. The release rate was also greatly affected by the medium composition. The other results are, m PBS of pH 7.4 only 33+1% of the peptide was released within 4 days, whereas, 53+2 and 61+0.95 were released in 1% BSA solution and serum respectively. The type of medium was found critical for the estimation of the in vivo release. From their investigations, it was concluded that the in vivo release kinetics of vapreotide pamoate form PLGA microspheres following administration to rats were qualitatively in good agreement with those obtained in vitro using serum as release medium and sterilization by g-irradiation had only a minor effect on the in vivo pharmacokinetics (124).
3.8 Alginate-poly-L-lysine microcapsules
Transplantation of islets of Langerhans as a means of treating insulin-dependent diabetes mellitus has become an important field of interest (125-127). However, tissue rejection and relapse of the initial autoimmune process have limited the success of this treatment. Immunoisolation of islets in semipermeable microcapsules has been proposed to prevent their immune destruction (128,129). Nevertheless, a pericapsular cellular reaction eventually develops around microencapsulated islets, inducing graft failure (130). Since empty microcapsule elicit a similar reaction (131), the reaction is not related to the presence of islets within the capsule but is, at least partially, caused by the capsule itself. Consequently, microcapsule biocompatibility appears to constitute a major impediment to successful microencapsulated islet transplantation.
Smaller microcapsules (<350 mm) offer many advantages over standard microcapsules (700 to 1500 mm), including reduced total implant volume, better insulin kinetics (132), improved cell oxygenation (133), and potential access to diverse implantation sites, such as the spleen and liver. A new electrostatic pulse system produces microcapsules of <350 mm in diameter (134,135) as compared with 700 to 1500 mm produced with the usual air-jet system. Lum et al (134) have suggested that these smaller microcapsules exhibit a higher degree of biocompatibility compared to standard microcapsules. However, no quantitative data or comparative studies have been published addressing this issue.
Previous investigations on alginate-poly-L-lysine microcapsule biocompatibility have focused mainly on intraperitoneal transplantation of microcapsulate islets into diabetic rats. The use of peritoneal implantation for biocompatibility studies is hindered by the fact that, in this site, microcapsules are unevenly distributed. Free floating microcapsules, which are easily recovered, are less likely to show pericapsular fibrosis than irretrievable microcapsules. The selection bias hinders quantitative evaluation of microcapsule biocompatibility. The low recovery of smaller microcapsules increases the selection bias. To overcome these methodological problems, Pariseau et al (136), developed and carefully validated a method for the in vivo comparative evaluation of microcapsule biocompatibility. This technique comprises implantation of microcapsules into rat epididymal fat pads, retrieval of fat pads after fixed time periods, and histological evaluations with the use of a fibrosis scove. Microcapsule recovery rate from this site was 99.6+0.75% (136). The pericapsular reaction is uniform within one fat pad and between fat pads, allowing random sampling and comparative studies (136).
Robitaille et al (137), reported investigation comparing the biocompatibility of microcapsules <350 mm in diameter made with an electrostatic pulse system to that of microcapsules 124+120 mm in diameter made with standard air-jet system, with the objectives;(1) to compare the biocompatibility of smaller and standard microcapsules while maintaining either equal implant volume or equal alginate content; and (2) to analyze the biocompatibility of smaller versus standard microcapsules with respect to the total implant surface exposed to the surroundings. To evaluate the biocompatibility, 200,1000,1120,1340, or 3000 of smaller microcapsules (<350 mm) or 20 standard microcapsules (1247+120 mm) were implanted into rat epididymal fat pads, retrieved after 2 weeks, and evaluated histologically. The average pericapsular reaction increased with the number of small microcapsules implanted (p<0.05; 3000 Vs 200, 300 Vs 1000 and 1000 Vs 200 microcapsules). At equal volume and alginate content, standard microcapsules caused a more intense fibrosis reaction than smaller microcapsules (p<0.05). In addition, 20 standard microcapsules elicited a stronger pericapsules (p<0.05) although the later represented a 3.4 fold larger total implant surface exposed. Finally, from their investigation it appears that the microcapsules of diameters <350 mm made with an electrostatic pulse system are more biocompatible than standard microcapsules (137).
3.9 Crosslinked chitosan microspheres
Procedure for the preparation of crosslinked chitosan microspheres coated with polysaccharide or lipid for intelligent drug delivery systems is reported Figure 10 (79).
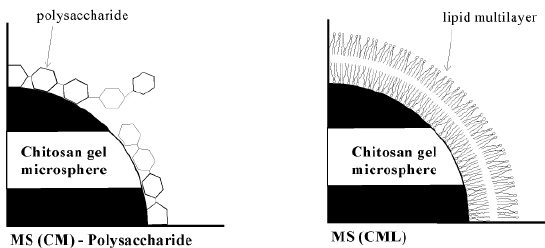
Figure 10 Theoretical structure of chitosan gel microsphere coated with polysaccharide or lipid
The microspheres were prepared with an inverse emulsion of 5-FU or its derivative solution of hydrochloric acid of chitosan in toluene containing SPAN 80. Chitosan was crosslinked with Schiff’s salt formation by adding glutaraldehyde toluene solution. At the same time, the amino derivatives of 5-FU were immobilized, obviously resulting in an increase in the amount of drug within the microspheres. The microspheres were coated with anionic polysaccharides (e.g. carboxymethylchitin, etc.) through a polyion complex formation reaction. In the case of lipid coated microsphere, the microspheres along with dipalmitoyl phosphalidyl choline (DPPC) were dispersed in chloroform. After evaporation of the solvent, microspheres were obtained coated with a DPPC lipid multilayer, which exhibited a transition temperature of a liquid crystal phase at 41.4 oC. The diameter range of microspheres was 250-300 nm with a narrow distribution. The stability of the dispersion was improved by coating the microsphere with anionic polysaccharide or a lipid multilayer Figure 11.
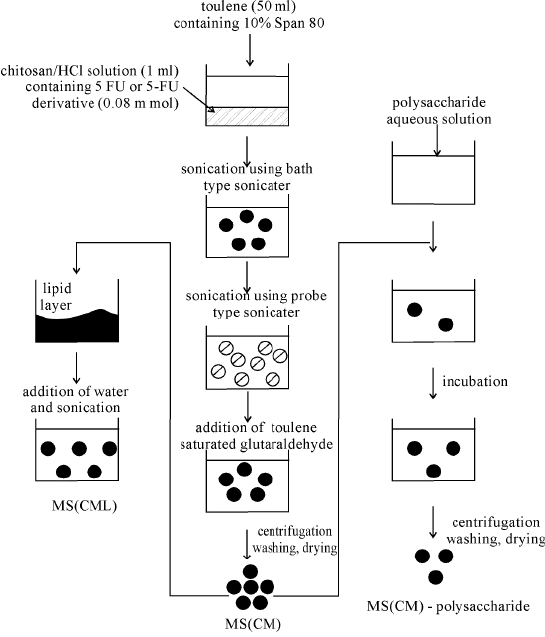
Figure 11 Preparation process of MS (CM), MS (CML) and MS(CML) and MC(CM) polysaccharide
A comparative study on the release of 5-FU and its derivatives from a polysaccharide coated microspheres MS (CM) was carried out in physiological saline at 37 oC. Data indicated that the 5-FU-release rate decreased in the order: free-5-FU > carboxymethyl type 5-FU > ester type 5-FU. The results revealed that the coating layers on the microspheres were effective barriers to 5-FU release.
The lipid mutilayers with a homogeneous composition generally show a transition of gel-liquid crystal. When the temperature is raised to 42 oC, which is higher than the phase transition of 41.4 oC, the release amount of 5-Fu increased, the amount of drug delivered decreased at 37 oC, which is lower than the transition temperature. Due to the improved recognition function of polysaccharide chains for animal cell membranes, it is reasonable to develop targeting delivery systems from polysaccharide coated microspheres, MS (CM).
3.10 Chitosan/gelatin microspheres
In their studies on pharmaceutical applications of chitin and chitosan, Yao and coworkers (138) reported chitosan/gelatin network polymer microspheres for controlled release of cimetidine. The drug loaded microspheres were prepared by dissolving chitosan, gelatin (1:1 by weight) and cimetidine in 5% acetic acid. A certain amount of Tween 80 and liquid paraffin at water to oil a ratio of 1:10 was added to the chitosan/gelatin mixture under agitation at 650 rev. per min at 30 oC. A suitable amount of 25% aqueous glutaraldehyde solution was added to the inverse emulsion and maintained for 2 h. Finally, the liquid paraffin was vaporized under vacuum to obtain microspheres.
The drug release studies were performed in hydrochloric acid solution (pH 1.0) and potassium dihydrogen phosphate (pH 7.8) buffer at ionic strength 0.1 m/L. A pH dependent pulsed-release behavior of the HPN matrix was observed (138). Moreover, the release rate can be controlled via the composition of the HPN and the degree of deacetylation of chitosan.
3.11 Crosslinked chitosan network beads with spacer groups
A novel technique for the preparation of pH sensitive beads of chitosan is reported as a part of the studies on controlled drug delivery applications of chitosan. Diclofenac sodium, Thyamine hydrochloride, chlorphenramine maleate and Isoniazid were used as model drugs (139-141). In these studies, widely used products in medical and pharmaceutical areas viz., glycine and polyethylene glycol were employed as spacer groups to enhance the flexibility of the polymer networks and influence the swelling behavior through macromolecular interactions. The procedure is based on adding drugs to chitosan solution and beads were prepared by simple coacervation (79). The swelling behavior, solubility, hydrolytic degradation and drug loading capacity of the beads were investigated (141,142). Effect of the crosslinker studied, by varying the amounts of crosslinker (142).
The beads exhibit high pH sensitivity. The swelling ratio of the beads at pH 2.0 is obviously higher than that at pH 7.4. This pH sensitive swelling is due to the transition of bead network between the collapsed and the expanded rates, which is related to ionization degree of amino groups on chitosan in different pH solutions. In both these systems, a near zero-order release is observed for about 4 days. The amounts and percent release in chitosan-PEG system is a bit higher, when compared to the chitosan-glycine system, due to the water diffusivity and pore forming properties of PEG. The effects of the amount of drug loaded and the crosslinking agent on the delivery profiles were reported as well (139-141).
3.12 1,5-diozepan-2-one (DXO) and D,L-dilactide (D,L-LA) microspheres.
The most successful class of degradable polymers so far have been aliphatic polyesters. The degradation takes place via hydrolysis of the ester linkages in the polymer backbone. These materials must be extensively tested and characterized since many of them are new structures. This is however, not sufficient. Equally important is the identification of degradation mechanisms and degradation products. Since high molecular weight polymers are seldom toxic, the toxicity and tissue response after the initial post operative period is related to the compounds formed during degradation.
The most important polymer on the market today is poly(lactic acid) (PLA) which upon degradation yields acetic acid, a natural metabolite in the human body. The formation of natural metabolites should be advantageous as the body has routes to eliminate them. Other commercial degradable materials are polyparadioxane, copolymer of glycolic acid and trimethylene carbonate which do not give natural metabolites when degraded. Their most important characteristic is probably the fact that the degradation products are harmless in the concentration present.
Albertsson and coworkers (143) carried out extensive research to develop polymers in which the polymer properties are altered for different applications. The predominant procedure is ring-opening polymerization which provides a way to achieve pure and well defined structures, They have utilized cyclic monomers of lactones, anhydrides, carbonates, ether-lactones, and specifically oxepan-2,7-dione (AA), b-propiolactone, e-caprolactone (e Cl), 1,5-dioxepan-2-one (DXO), dilactide, and 1,3-dioxan-2-one (TMC). The work involved the synthesis of monomers not commercially available, studies of polymerization to form homopolymers, statistical and block copolymers, development of crosslinked polymers and polymer blends, surface modification in some cases, and characterization of the materials formed. The characterization is carried out with respect to the chemical composition and both chemical and physical structure, the degradation behavior in vitro and in vivo, and in some cases the ability to release drug components from microspheres prepared from the polymers.
Copolymers of 1,5-dioxepan-2-one (DXO) and D, L-dilactide (D,L-LA) were synthesized in different molar concentration, in their recent attempts (144). 1H-NMR was used to determine the molar composition of the copolymers, and DSC was used to determine the glass transition and melting temperatures. All the polymers were amorphous with a glass transition varying between -36 oC [poly(DXO)] to +55 oC [poly(D, L-LA)]. In vitro hydrolysis studies on the copolymers showed degradation times up to 250 days. Copolymers of 1,3-dioxan-2-one (TMC) and e-caprolactone (e Cl) were also studied (144). Conversion studies were performed, and both monomers separately showed almost the reactivity. Poly(e-caprolactone) seemed to be more sensitive to transesterification at elevated temperatures than poly(trimethylene carbonate). Using DSC, melting endotherms were seen in molar compositions with a CL content as low as 65% which indicates a block structure. Microspheres intended for drug delivery were prepared from poly(TMC-co-CL), poly(adipic anhydride) and poly(lactide-co-glycolide) (144). SEM studies showed that the microsphere morphology concentration dependent at the time of preparation and on the choice of polymer. The drug release profiles showed dependence on the polymer degradation behavior and on the water penetration into the microsphere (144).
3.13 Triglyceride lipospheres
Liquid drug-delivery systems, which do not require surgical implantation, may use vehicles made of microparticulates or colloidal carriers composed of lipids, carbohydrates or synthetic polymer matrices. Liposomes, the most widely studied of these vesicles, can be formulated to include a variety of compositions and structures that are potentially non-toxic, degradable and nonimmunogenic. To produce a long acting local anesthetic effect, vesicles have been used to entrap dibucaine (145), methoxyflurane (146), tetracaine (147) and lidocaine (148) using formulations with polylactic acid, lecithin, iophendylate and phosphatidylcholine and cholesterol, respectively. With varying degrees of success, these treatments have provided neural blokade for periods far outlasting, which is produced by any drug given alone.
Masters and Domb (149) reported on an injectable drug delivery system that uses liposomes (150) to release the local anesthetic, bupivacaine, from a liposomal matrix that is both biodegradable and biocompatible to produce sustained local anesthetic blockade (SLAB). Bupivacaine due to its minimum vasodilating properties was preferred to other local anesthetics (e.g., lidocaine) allowing the released drug to remain at the site of injection longer (151). Lipospheres are an aqueous microdispersion of water insoluble, spherical microparticles (0.2 to 100 mm in diameter), each consisting of a solid core of hydrophobic triglycerides and drug particles that are embedded with phospholipids on the surface. The in vivo studies with Liposheres have shown that a single bolus injection can deliver antibiotics and anti-inflammatory agents for 3 to 5 days (152) and also, control the delivery of vaccines (153,154). Recent reports were of bupivacaine-liposphere formulation, which produced 1-3 days reversible sensory and motor SLAB when applied directly to the rat sciatic nerve (149). The particle size of the liposheres was in between 5 and 15 mm, with over 90% surface phospholipid. Lipospheres released bupivacaine over two days under ideal sink conditions. Liposphere nerve application produced dose-dependent and reversible block. Sustained local anesthetic block (SLAB) was observed for 1-3 days in various in vivo tests: (a) Hind paw withdrawal latency to noxious heat was increased over 50% for 96h period after application of 3.6% or 5.6% bupivacaine-lipospheres. The 3.6% and 5.6% doses were estimated to release bupivacaine at 200 and 311 mg drug/h, respectively based on release spanning 72h. Application of 1.6% bupivacaine-lipospheres increased withdraw latency 25-250% but for only a 24h duration, (b) similarly, vocalization threshold to hind paw stimulation was increased 25-50% for 72h following application of 3.6% bupivacaine-lipospheres; (c) finally concluded that, sensory blockade outlasted or equaled corresponding motor block duration for all liposphere durg dosages (149).
3.14 Glutamate and TRH microspheres
L-glutamate is the principal excitatory neurotransmitter in the mammalian central nervous system (155) and has been shown to stimulate trigeminal motoneurons within the trigeminal motor nucleus in acute, short-term physiologic studies (156). Since trigeminal motoneurons innervate the muscles of mastication and activity patterns (EMG) of those muscles directly affect the growth/development of the carnifacial skeleton by biomechanical forces produced (157), Byrd and coworkers (158,159), tried and successfully used gultamate-impregnated microspheres to stimulate trigeminal motoneurons in situ within the brainstem of young rats to produce skeletal alterations. The underlying premise was that increased delivery of glutamate in proximity to trigeminal motoneurons would increase activity of both those motoneurons and the masticatory muscles they supply. Indirect physiologic evidence for this premise was provided by the presence of (1) more pronounced implantside wear facets on the mandibular incisors in rats with gultamate-microsphere implants, and (2) deviation of their facial skeletons toward the implant side (158,159). The microspheres used were of biodegradable, polyanhydride construction and released gultamate at a controlled rate within the intact rat. The use of biodegradable polyanhydride microspheres as drug-carrier matrices (160) is an effective method for delivery of long-term release of neuroactive substances to a specific locus within the CNS with little risk of infection (159).
The tripeptide TRH (thyrotropin-releasing hormone) has been confirmed as an important neurotransmitter/neuromodulator within the brain stem region of the CNS (161-165). Trigeminal motoneurons are highly immunoreactive for TRH (163) and also that TRH actually increases the excitability of other brain stem motoneurons in vitro (165). TRH therefore proved to be useful to increase activity levels of trigeminal motoneurons in vivo. Byrd et al (158), investigated in the chronic, long-term effects of administering TRH in proximity to trigeminal motoneurons in vivo, and comparing any carniofacial sequelae with those effected by glutamate microspheres.
Recently, Byrd et al (166) investigated the sequelae of sustained, in vivo delivery of two important neurotransmitter substances, glutamate and TRH, upon carniofacial growth and development. In their studies, the relative effects of glutamate and TRH microspheres stereotactically placed in proximity to trigeminal motoneurons within the trigeminal motoneurons were compared. Stereoactive neurosurgery at 35 days was conducted for 4 experimental groups comprising 10 male Sprague-Dawley rats a group. The data was collected after killing 5 rats of each group at 14 and 21 days. Histology revealed that implants were clustered in the pontine reticular formation, close to ventrolateral tegmental nucleus. Both glutamate and TRH rats had implant-side deviation of their facial skeleton and snout regions 4x2 ANOVA and post hoc t-tests revealed significant (p < 0.05,0.01) differences between groups and sides for motoneuron count, muscle weight, and osteometric data.
3.15 Polyelectrolyte complexes of sodium alginate chitosan
Polyelectrolyte complexes (PECs) are formed by the reaction of a polyelectrolyte with an oppositely charged polyelectrolyte in an aqueous solution. Polysaccharides, which have bulky pyranose rings and highly stereoregular configuration in their rigid, linear backbone chains, have been frequently studied (167). PECs have numerous applications such as membranes, antistatic coatings, environmental sensors, and chemical detectors, medical prosthetic materials etc (168). Among these, their wide use as membranes for dialysis, ultrafiltration, and other solute separation processes are of special interest and also made it possible for the use in microcapsule membranes. Microcapsules can be used for mammalian cell culture and the controlled release of drugs, vaccines, antibiotics, and hormones (168-170), To prevent the loss of encapsulated material, the microcapsules should be coated with another polymer that forms a membrane at the bead surface. The most well known and promising system is the encapsulation of alginate beads with poly-L-lysine (PLL). Because this system has a limitation due to the high cost of PLL, other systems such as alginate beads coated with chitosan or its derivatives have been developed (83,84). Few results have been reported about the formation of PECs of alginate with chitosan under acidic condition. Although alginate/chitosan microcapsules have been studied a lot, the studies have been limited in a narrow pH region due to the solubility of chitosan.
Lee et al (171) described a new procedure to overcome the solubility of chitosan. In this procedure, chitosan was heterogeneously deacetylated with a 47% sodium hydroxide solution and followed by a homogeneous reacetylation with acetic anhydrides to control the N-acetyl content of the chitosan having a similar molecular weight. The chitosan having different degrees of N-deacetylation were complexed with sodium alginate, and the formation behavior of polyelectrolyte complexes (PECs) was examined by the viscometry in various pH ranges. The maximum mixing ratio (Rmax) increased with a decrease in the degree of N-acetylation of the chitosan at the same pH, and with a decrease in pH at the same degree of N-acetylation. Similarly, N-acylated chitosans were also prepared. The N-acyl chitosans scarcely affected the formation behavior of PECs with sodium alginates. For the application of PECs produced, the microencapsulation of a drug was performed and the release property of drug was tested. The microcapsules were prepared in one step by the extrusion of a solution of a guaifenesin and sodium alginate into a solution containing calcium chloride and chitosan through inter-polymeric ionic interactions. The drug release during the drug-loaded microcapsules storage in saline was pH dependent, where the microcapsules were formed and the kind of N-acyl groups introduced to the chitosan. The microcapsules prepared at pH 4.8 showed a minimum release rate and the release rate varied with the pH due to the loop formation of backbone chains of polyelectrolytes. The N-acyl groups introduced to the chitosan enhanced the release rate remarkably.
3.16 Polypeptide microcapsules
A number of synthetic polypeptides have been investigated for medical applications such as biodegradable suture, artificial skin substitutes and sustained release devices. The rate of degradation of synthetic polypeptides can be controlled by a proper selection of amino acid components. Recent reports revealed, microcapsules prepared from [Glu(OMe)]m (Sar)n (m=21, n=19) and [Lys(z)]m (Sar)n (m=27, n=15), and were chemically modified to obtain a pH-responsive releasing membranes. One membrane was prepared by partially deprotecting the ester groups of [Glu(OMe)m (Sar)n. The other membrane was prepared by connecting of poly(Glu) to side chain amino groups that were generated by a partial deprotection of [Lys(z)]m (Sar)n. In the later stages, two types of polypeptide microcapsules were prepared; Glu residues in the main chain, and Glu residues in the graft chains on the positively charged main chain. Both microcapsules showed pH-responsive release of FITC-dextran encapsulated in the microcapsules. The release rate was observed to be slower in the medium at pH3.0 than pH 7.5. From optical microscope observation, it appears that partially deblocked [Glu(OMe)]m (Sar)n microcapsules swelled more at pH 7.5 than at pH 3.0; might be due to enhanced permeation through the polypeptide membrane at pH 7.5. However, the authors observed a little change in the shape of poly(Glu)-grafted [Lys(z)]m (Sar)n microcapsules, by changing pH of the medium. It is suggested that ion-pairing between carboxylate groups of poly(Glu) and ammonium groups of Lys acts as crosslinking to give the shape stability (172).
3.16 Albumin microspheres
Albumin is an attractive macromolecular carrier and widely used to prepare microspheres and microcapsules, due to its availability in pure form and its biodegradability, nontoxicity and nonimmmunogenicity (173). A number of studies have shown that albumin accumulates in solid tumors (174,175) making it a potential macromolecular carrier for the site-directed delivery of antitumor drugs. Recently, Katti and Krishnamurti (176) prepared albumin microspheres by suspension crosslinking in the absence of any surfactant using paraffin oil as the dispersion medium and formaldehyde as the crosslinking agent. They characterized the microspheres by SEM and found to be spherical having a particle size distribution in the range of 50-400 mm. A preliminary drug release study of chlorothiazide in vitro indicated a diffusion controlled release of drug. Authors claimed that this method is simple, cost effective and moreover, promising technique for the large scale manufacturing (176).
4. Acknowledgement
The author is grateful to Council of Scientific and Industrial Research (CSIR), Ministry of Human Resource Development Group, Govt. of India, New Delhi) for financial assistance.
References
-
Diepold, R., Kreuter, J., Guggenbuhl, P. and Robinson, J. R., Distribution of poly-hexyl-2-cyano-[3-14C] acrylate nanoparticles in healthy and chronically inflamed rabbit eyes. Int. J. Pharm., 54: 149-153, 1989.
-
Illum, L., Wright, J. and Davis, S. S., Targetting of microspheres to sites of inflammation. Int. J. Pharm., 52: 221-224, 1989.
-
Alpar, H. O., Field, W. N., Hyde, R. and Lewis, D. A., The transport of microspheres from the gastro-intestinal tract to inflammatory air pouches in the rat. J. Pharm. Pharmacol., 41: 194-196, 1989.
-
Couvreur, P. Kante, B., Grislain, L., Roland, M. and Speiser, P., Toxicity of polyalkylcyanoacrylate nanoparticles II: Doxorubicin-loaded nanoparticles. J. Pharm. Sci., 71: 790-792, 1982
-
Couvreur, Grislain, L., Lenaert, V., Brasseur, F., Guiot, P. and Biernacki, A., Biodegradable polymeric nanoparticles as drug carrier for antitumor agents. In Guiot, P. and Couvreur, P., (Eds.), Polymeric Nanoparticles and Microspheres, CRC Press, Boca raton, 1986, pp. 27-93.
-
Labhasetwar, V., Song, C. and Levy, R. J., Nanoparticle drug delievry systems. Adv. Drug. Del. Rev., 24: 63-85, 1997.
-
Jagur-Grodzinski, J., Biomedical application of functional polymers. React. Funct. Polym., 39: 99-138, 1999.
-
Kwon, G.S. and Okano, T., Polymeric micelles as new drug carriers. Adv. Drug Del. Rev., 21: 107-116 1996.
-
Scholz, C., Iijima M., Nagasaki Y. and Kataoka K., A novel reactive polymeric micelle with aldehyde groups on its surface. Macromolecules, 26: 7295-7297, 1995.
-
La, S. B., Okano, T. and Kataoka, K., Preparation and characterization of the micelle-forming polymeric drug iodomethacin-incorporated poly(ethylene oxide)-poly(b-benzyl-L-aspartate) block polymer micelles. J. Pham. Sci., 85: 85-90, 1996.
-
Zhang, X. and Burt, H. M., Diblock copolymers of poly(DL-lactide)-block-methoxy poly(ethylene glycol) as mecillar carrier of taxol. Pharm. Res., 12: S265, 1995.
-
Dunn, S. E., Coombes, A. G. A., Garnett, M. C., Davis, S. S., Davis, M. C. and Illum, L. J., In vitro cell interaction and in vivo biodistribution of poly(lactide-co-glycolide) nanosphere surface modified poloxamer and poloxamin copolymers. J. Controlled Release, 44: 65-76, 1997.
-
Fessi, H. et al., FR 2608988 and FR 2608942, July 1988.
-
Moghimi, S. M., Muir, I. S., Illum, L., Davis, S. S., and Kolb Bachofen, V., Coating particles with a block co-polymer (poloxamine-908) suppresses opsonization but permits the activity of dysopsonins in the serum. B.B.A., 1179: 157-165, 1993.
-
Allock, H. R. and Chang, J.-Y., Poly(organophosphazenes) with oligopeptides as side groups: prospective biomaterials. Macromolecules, 24: 993-999, 1991.
-
Allock, H. R., Ducher, S. R., and Scopelianos, A. G., Poly[(amino acid esters) phosphazene] as substrates for the controlled release of small molecules. Biomaterials, 15: 563-569, 1994.
-
Allock, H. R., Fuller, T. J., Mack, D. P., Matsumura, K. and Smeltz K. M., Synthesis of [poly(amino acid alkylester) phosphazenes]. Macromolecules, 10: 824-830, 1997.
-
Vandorpe, J., Schacht, E. et al, [Poly(organophosphazene)] nanoparticles surface modified with polyethylene oxide. Biotechnol. Bioeng., 52: 89-95, 1996.
-
Vandorpe, J., Schacht, E., et al., Long circulating biodegradabale poly(phosphazene) nanoparticles surface modified with poly(phosphazene)-polyethylene oxide co-polymer. Biomaterials, 18: 1147-1152, 1997.
-
Gref, R., Minamitake Y., et al., Biodegradable long circulating nanospheres. Science, 263: 1600-1603, 1994.
-
Gref, R., Domb, A. et al., The controlled intraveneous delivery of drugs using PEG-coated sterically stabilized nanospheres. Adv. Drug Del. Rev., 16: 215-233, 1995.
-
Hrkach, J. S., Peracchia, M. T., Domb, A., Lotan, N. and Langer, R., Nanotechnology for biomaterials engineering: structural characterization of amphiphilic polymeric nanoparticles by 1H NMR spectroscopy. Biomaterials, 18: 27-30, 1997.
-
Coombes, A. G. A., Tasker, S., Lindblad, M., Holmgreen, K., Hoste, V., et al., Biodegradable polymeric microparticles for drug delivery and vaccine formulation: the surface attachment of hydrophilic species using the concept of poly(ethylene glycol) anchoring segments. Biomaterials, 18: 1153-1161, 1997.
-
Kreuter, J., Recent advances in naoparticles and nanospheres. in Hincal, A. A. and Kas, H. S. (Eds.), Biomedical Science and Technology, Plenum Press, New York, 1998, pp. 31-39.
-
Levy, J. A., Pathogenesis of human immunodeficiency virus infection. Microbiol. Rev., 57: 183-289, 1993.
-
Weiss, R. A., How does HIV cause AIDS? Science, 260: 1273-1279, 1993.
-
Gartner, S., et al., The role of mononeuclear phagocytes in HTLV-III/LAV infection. Science, 223: 215-219, 1986.
-
Kuhnel, H., Markovitis, P., Markovitis, D. M., Kalpan, M. H., et al., Molecular cloning of two West African human immunodeficiency virus type 2 isolates that replicate well in macrophages: a Gambian isolate, from a patient with neurologic acquired immunodeficiency syndrome and a highly divergent Ghaninan isolate. Proc. Natl. Acad. Sci., USA, 86: 2383-2387, 1989.
-
Briesen, H. von, Andreesen, R., Esser, R., Brugger, W., et al., Infection of monocytes/macrophages by HIV in vitro. Res. Virol., 141: 225-231, 1990.
-
Esser, R., Briesen, H. von, Brugger, W., Ceska, M. et al., Secretory repertoire of HIV-infected human monocytes/macrophages. Pathobiology, 59: 219-222, 1991.
-
Gendelman, H. E., Lipton, S. A., Tardieu, M., Bukrinsky, M. I., et al., The neuropathogenesis of HIV-I infection. J. Lenukocyte Biol., 56: 389-398, 1994.
-
Bender, A. R., Brisen, H.von, Kreuter, J., Duncan, I. B., and Rubsamen, H., Efficiency of nanoparticles as a carrier system for antiviral agents in human immunodeficiency virus-infected human manocytes/macrophages in vitro. Antimicrob. Agents Chemother. 40: 1467-1471, 1996.
-
McGann, K. A., Collmann, R., Kolson, D. L., Gonzalez-Scarano, F., et al, Human immunodeficiency virus-I causes productive infections of macrophages in primary placental cell cultures. J. Infect. Dir., 169: 746-753, 1994.
-
Milman, G. and Sharma O., Mechanism of HIV/SIV mucosal transmission. AIDS Res. Hum. Retrovisures, 10: 1305-1312, 1994.
-
Vanct-Wout, A. B., Kootstra, N. A., Mulder-Kampinga, G. A., et al., Macrophage-tropic variants initiate human immunodeficiency virus type I infections after sexual, parenteral and vertical transmission. J. clin. Invest., 94: 2060-2067, 1994.
-
Schafer, V., Briesen, H. von, Andreesen, R., Steffan. A.-M., et al., How to get antiviral into HIV-infected cells? Phagocytosis of nanoparticles by human macrophages. Microbiologist, 2: 117-121, 1991.
-
Schafer, V., Briesen, H. von, Andreesen, R., Steffan. A.-M., et al., Phagocytosis of nanoparticles by human immunodeficiency virus (HIV)-infected macrophages: A possibility for antiviral drug targeting. Pharm. Res., 9: 541-546, 1992.
-
Bender, A. Schafer, V., Steffan, A.-M., Royer, C. et al., Inhibition of HIV in vitro by antiviral drug-targeting using nanoparticles. Res., Virol., 145: 215-220, 1994.
-
Lobenberg, R. and Kreuter, J., Macrophage targeting of azidothymidine: A promising strategy for AIDS therapy. AIDS Res. Human Retrovir., 12: 1709-1715, 1996.
-
Lobenberg, R., Mass, J. and Kreuter, J., Improved body distribution of 14C-labelled AZT bound to nanoparticle in rats determined by radioluminograpgy. J.Drug Targeting, 14: 1247-1253, 1999.
-
Aprahamian, M., Michel C., Humbert W., Devissaguet J.P. and Damge, C., Transmucosal passage of polyalkylcyanoacrylate nanocapsules as new drug carrier for small intestine. Biol. Cell, 61: 69-76, 1987.
-
Damge, C., Michel C., Aprahamian, M. and Couvreur, P., New approaches for oral administration of insulin with polyalkylcyanoacrylate nanocapsules as a drug carrier. Diabetes, 37: 246-251, 1988.
-
Damage, C., Michel, C., Aprahamian, M., Couvreur, P. and Devissaguet, J. P., Nanocapsules as carrier for oral peptide delivery. J. Control Release, 13: 233-239, 1990.
-
Damage, C., Hillaire-Buys, D., Puech, R., Hoeltzel, A., Michel, C., and Ribes, G., Effect of orally administered insulin nanocapsules in normal and diabetic dogs. Diab. Nutr. Metab., 8: 3-9, 1995.
-
Alkhouri Fallouh, N., Roblot, Treupel, L., Fessi, H., et al, Development of a new process for manufacture for polyisobutylcyanoacrylate nanocapsules. Int. J. Pharm., 28: 125-132, 1986.
-
Leonard, F. Kulkarni, R.K., Brandes, G., Nelson, J. and Cameron, J., Synthesis and degradation of poly(alkyl-a-cyanoacrylates). J. Appl. Polym., Sci., 10: 259-272, 1996.
-
Damge, C., Vranckx, H., Balschmidt, P. and Couvreur, P., Poly(alkylcyanoacrylate) nanospheres for oral administration of insulin. J. Pharm. Sci., 86: 1403-1409, 1997.
-
Aboubakar, M., Puisieux, F., Couvreur, P. and Vauthier, C., Physico-chemical characterization of insulin-loaded poly(isobutylcyanoacrylate) nanocapsules obtained by interfacial polymerization. Int. J. Pharm., 183: 63-66, 1999.
-
Brannon-Peppas, L., Recent advances on the use of biodegradable microparticles and nanoparticles in controlled drug delivery. Int. J. Pharm., 116: 1-9, 1995.
-
Langer, K., Stieneker, F., Lambrecht, G., Mutschler, E. and Kreuter, J., Methylmethacrylate sulfopropylmethacrylate copolymer nanoparticles for drug delievry. Part II: arecaidine propargyl ester and pilocarpine loading in vitro release. Int. J. Pharm., 158: 211-217, 1997.
-
Maruyama, A., Ishihara, T. Kim, J.-S., Kim, S.-W. and Akaike, T., Nanoparticle DNA carrier with poly(L-lysine) grafted polysaccharide copolymer and poly(D,L-lactic acid). Bioconjugate Chem., 8: 735-742, 1997.
-
Paul, M., Fessi, H., Laatiris-Boulald, Y., et al., Pentamidine-loaded poly(D,L-lactide) nanoparticles: physicochemical properties and stability work. Int. J. Pharm., 159: 223-232, 1997.
-
Coffin, M. D. and McGinity, J. W., Biodegradable pseudolatexes: The chemical stability of poly(D,L-Lactide) and poly(e-Caprolactone) nanoparticles in aqueous media. Pharm. Res., 9: 200-205, 1992.
-
Lee Ray, A. M., Vert, V., Gautier, J. C. and Benoit, J. P., Fate of [14C] poly(D,L-lactide-co-glycolide) nanoparticles after intravenous and oral administration to mice. Int. J.Pharm., 106: 201-211, 1994.
-
Zhang, Q., Liao, G., Wei, D., and Nagai, T., Increase in gentamicin uptake by cultured mouse peritoneal macrophages and rat hepatocytes by its binding to poly-butylcyanoacrylate nanparticles. Int. J. Pharm., 164: 21-27, 1998.
-
Masson, V., Maurin, F., Devissaguet, J. P. and Fessi, H., Stability of poly(e-Caprolactone) nanospheres in sterile aqueous media. Int. J. Pharm, 139: 113-123, 1996.
-
Gao, Z. and Eisenberg, A., A model of micellization for block copolymers in solutions. Macromolecules, 26: 7353-7360, 1993.
-
Hruska, Z., Kwon, G. S., Yokoyama, M., Okano, T. and Sakurai, Y., Synthesis and purification of a poly(ethylene oxide)-poly(g-benzyl-L-glutamate)diblock copolymer bearing tyrosine units at the block junction. J.Control Release, 34: 1333-1335, 1993.
-
Yokoyama, M., Okano, T., Sakurai, Y., Ekimoto, H., Shibazaki, C. and Kataoka, K., Toxicity and antitumor activity against solid tumors of micelle forming polymeric anticancer drug and its extremely long circulation in blood. Cancer Res., 51: 3229-3236, 1991.
-
Kataoka, K., Kwon, G. S., Yokoyama, M., Okano, T. and Sakurai, Y., Block copolymer micelles as vehicles for drug delivery. J. Control. Release, 24: 119-132, 1993.
-
Kwon, G. S., Suwa, S., Yokoyana, M. Okano, T. Okano, Sakuri Y. and Kataoka, K., Enhanced tumor accumulation and prolonged circulation time of micelle-forming poly(ethylene oxide- aspartate) block copolymer-adriamycin conjugates. J. Control. Release, 29: 17-23, 1994.
-
Oh, I., Lee, K., Kwon, H.-Y., Lee, Y.-B. Shin, S.-C., Cho, C.-S. and Kim, C.-K., Release of adriamycin from poly(g-benzyl-L-glutamate)/poly(ethylene oxide) nanoparticles. Int. J. Pharm., 181: 107-115, 1999.
-
Cho. C.-S., Kim, S.-W. and Komoto, T., Synthesis and structural study on an ABA block copolymer consisting of poly(g-benzyl-L-glutamate) as the A block and poly(ethylene oxide) as the B block. Macromol. Rapid Commun., 191: 981-991, 1997.
-
Jeong, Y. I, Cheon, J. B., Kim, S.-H., Nah, J. W., Lee, Y.-M., Chlonazepam release from core-shell type nanoparticles in vitro. J. Control Release, 51: 169-178, 1998.
-
Amiji, M. and Park, K., In Polymers of biological Significance, Shalaby, S. W., Ikada Y. Langer R. and Welliams J. (Eds.), ACS symposium series 540, washington, DC, 1994.
-
Lehr, C. M., Boustra, J. A., Schacht, E. H. and Junginer, H. E., In vitro evaluation of mucoadhesive properties of chitosan and some other natural polymers. Int J. Pharm., 78: 43-48, 1992.
-
Pornsak, S., Delivery systems for peptide and protein drugs. Warasan Phesatchasat, 23: 1- 12, 1996.
-
Calvo, P., Remunan-Lopez, C., Vila-Jato, J. L. and Alonso, M. J., Novel hydrophilic chitosan-poly(ethylene oxide) nanoparticles as protein carriers. J.Appl.Polym.Sci., 63: 125- 132, 1997.
-
Chang, J. and Zhongguo, S. Y., Application of nanoparticles in controlled release of anticancer drugs: preparation and evaluation in vitro of methotrexate-o-carboxymethylate chitosan nanoparticles. Chinese J. Biomed.Eng., 15: 102-106, 1996.
-
Kaplun, A. P., Son, L. B., Krasnopolsky, Y. M. and Shvets, V. I., Liposomes and other nanoparticles as drug delivery systems. Vopr.Med.Khim., 45: 3-12, 1999.
-
Schwarz, C. and Mehnert, W., Solid lipid nanoparticles (SLNs) for controlled drug delivery II. Drug incorporation and physicochemical characterization. J. Microencapsulation, 16: 205-213, 1999. (References therein)
-
Kim, Y. S. and Kim, K. S., Particle size distribution, drug loading capacity and release profiles of solid lipid nanoparticles of phenylpropionic acids. Yakehe Hakhoechi (Korean), 28: 249-255, 1998.
-
Nishimura, K., Nishimura, S., Seo, H., Nishi, N., Tokura, S. and Azuma, I., Macrophage activation with multiporous beads prepared from partially deacetylated chitin. J. Biomed. Mater. Res., 20: 1359-1372, 1986.
-
Ida, G., Monica, C., Annalia, A., Bice, C. and Luisa, M., Influence of glutaraldehyde on drug release and mucoadhesive properties of chitosan microspheres. Carbohydr.Polym., 36: 81-88, 1998.
-
Hayashi, T. and Ikada, Y., Protease immobilization onto porous chitosan beads, J. Appl. Polym. Sci., 42: 85-92, 1991.
-
Chandy, T. and Sharma, C. P., Chitosan beads and granules for oral sustained delivery of nifedipine: in vitro Studies. Biomaterials, 13: 949-952, 1992.
-
Chandy, T. and Sharma, C. P., Chitosan matrix for oral sustained delivery of ampicillin. Biomaterials, 14: 939-944, 1993.
-
Chandy, T., Sharma, C. P. and Sunny, M. C., Inhibition of platelet adhesion to glow discharge modified surfaces. J. Biomat. Appln., 1: 533-552, 1987.
-
Yao, K.-D., Peng, T., Yu, J.-J., Xu, M.-X. and Goosen, M. F. A., Microcapsules/ Microspheres related to chitosan. J. M. S.-Rev. Macromol. Chem. Phys., C35: 155-180, 1995.
-
Uhrich, K. E., Cannizzaro, S. M., Langer, R. S. and Shakesheff, K. M., Polymeric systems for controlled drug release. Chem.Rev., 99: 3181-3198, 1999.
-
Wheatley, M. A., Chang, M., Park, E. and Langer, R., Coated alginate microspheres factors influincing the controlled delivery of macromolecules. J.Appl.Polym.Sci., 43: 2123-2135, 1991.
-
Murata, Y., Toniwa, S., Miyamoto, E. and Kawashima, S., Preparation of alginate gel beads containing chitosan salt and their function. Int.J.Pharm., 176: 265-268, 1999.
-
Dunn, E. J., Zhang, X., Sun, D. and Goosen, M. F. A., Synthesis of N-(aminoalkyl) chitosan for microcapsules. J.Appl.Polym.Sci., 50: 353-365, 1993.
-
Huguet, M. L., Groboillot, A., Neufeld, R. J., Poncelet, D. and Dellacherie, E., Hemoglobin encapsulation in chitosan/calcium alginate beads. J. Appl. Polym. Sci., 51: 1427-1432, 1994.
-
Huguet, M. L. and Dellacherie, E., Calcium alginate beads coated with chitosan: effect of structure encapsulated materials on their release, Process Biochemistry, 31: 745-751, 1996.
-
Huguet, M. L., Neufeld, R. J. and Dellacherie, E., Calcium alginate beads coated with polycationic polymers: comparison of chitosan and DEAE-dextran, Process Biochemistry, 31: 347-353, 1996.
-
Seig, J. W. and Robinson, J. R., Vehicle effects on occular drug bioavailability. I. Evaluation of fluorometholone. J.Pharm.Sci., 64: 931-936, 1975.
-
Lee, V. H. L. and Robinson, J. R., Review: topical occular drug delivery: recent developments and future challenges. J.Ocul.Pharmacol., 2: 67-108, 1986.
-
Albertsson, A.-C., Carlfors, J. and Sturesson, C., Preparation and characterization of poly(adipic anhydride) microspheres for ocular drug delivery. J.Appl.Polym.Sci., 62: 695-705, 1996.
-
Kang, K. S., Veeder, G. T., Mirrasoul, P. J., Kaneko, T. and Cottrell, I. W., Agar-like polysaccharide produced by pseudomonas species: production and basic properties. Appl. Environ. Microbiol., 43: 1086-1091, 1982.
-
Doner, L. W. and Douds, B. D., Purification of commercial gellan to mono gellan cation salts results in acute modification of solution and gel forming properties. Carbohydr. Res., 273: 225-233, 1995.
-
Jansson, P. E., Lindberg, B. and Sandford, P. A., Structural studies of gellan gum, an extra cellular polysaccharide elaborated by pseudomonas elodea. Carbohydr. Res., 124: 135-139, 1983.
-
O'Neill, M. A., Selvendran, R. R. and Morris, N. J., Structure of the acidic extra cellular gelling polysaccharide produced by pseudomonas elodea. Carbohydr. Res., 124: 123-133, 1983.
-
Kuo, M. S., Mort, A. J. and Dell, A., Identification and location of L-glycerate an unusual acyl substituent in gellan gum. Carbohydr. Res., 156: 173-187, 1986.
-
Chandrasekaran, R., Radha, A. and Thailembal, V. G., Role of potassium ions, acetyl and L-glyceryl groups in native gellan double helix: an X-ray study. Carbohydr. Res., 224: 1-17, 1992.
-
Sanderson, G. R. and Clark, R. C., Gellan gum. Food Technol., 37: 62-70, 1983.
-
Milas, M., Shi, X. and Rinando, M., On the physicochemical properties of gellan gum. Biopolymers, 30: 451-464, 1990.
-
Grasdalen, H. and Smidsrod, O., Gelation of gellan gum. Carbohydr. Polym., 7: 371-393, 1987.
-
Deasy, P. B. and Quigley, J., Rheological evaluation of deacetylated gellan gum (gelrite) for pharmaceutical use. Int. J. Pharm., 73: 117-123, 1991.
-
Anderson, D. M. W., Brydon, W. G. and Eastwood, M. A., The dietary effect of gellan gums in humans. Food. Addit. Contam., 5: 237-249, 1988.
-
Shungu, D., Valiant, M., Tutlane, V., Weinberg, E., et al., Gelrite as an agar substitute in bacteriological media. Appl. Environ. Microbiol., 46: 840-847, 1983.
-
Harris, J. E., Gelrite as an agar substitute for the cultivation of mesophilic methanocaterium and methanobrevibater species. Appl. Environ. Microbiol, 50: 1107-1109, 1985.
-
Colegrove, G. T., Agricultural application of microbial polysaccharides. Ind. Eng. Chem. Prod. Res. Dev., 22:456-460, 1983.
-
Rozier, A., Mazuel, C., Grove, J., and Plazonnet, B., GelriteÒ a novel, ion activated in situ gelling polymer for ophthalmic vehicle. Effect on bioavailability of timolol. Int. J. Pharm., 57: 163-168, 1989.
-
Rozier, A., Mazuel, C., Grove, J., and Plazonnet, B., Functionality testing of gellan gum, a polymeric exciepent material for ophthalmic dosage forms. Int. J. Pharm., 153: 191-198, 1997.
-
Greaver, J. L., Wilion, C. G., Rozier, A., Grove, J. and Plazonnet, B., Scintigraphic assessment of an ophthalmic gelling vehicle in man and rabbit. Curr. Eye Res., 9: 415-420, 1990.
-
Sanzgiri, Y. D., Maschi, S., Crescenzi, V., Callegaro, L., et al., Gellan-based systems for ophthalmic sustained delivery of methylprednisolone. J. Control. Release, 26: 195-201, 1993.
-
Chilvers, G. R. and Morris, V. J., Coacervation of gelatin-gellan gum mictures and their use in microencapsulation. Carbohydr. Polym., 7: 111-120, 1987.
-
Alhaique, F., Carafa, M., Coviello, T., Murtas, E., et al., Gellan in sustain release formulations: release of thephylline from beads and gel capsules. Proc. Control Release Soc., 22: 286-287, 1995.
-
Alhaique, F., Carafa, M., Coviello, T., Murtas, E., et al., Gellan in sustain release formulations: preparation of gel capsules and release studies. J. Control Release, 42: 1981-1986, 1996.
-
Santucci, E., Alhaique, F., Carafa, M., Coviello, T., et al., Gellan for the formulation of sustained delivery beads. J. Control. Release, 42: 157-164, 1996.
-
Kedzierewicz, F., Lombry, C., Rios, R., Hoffman and M., Maincent, P., Effect of the formulation on the in vitro release of propranolol from gellan beads. Int. J. Pharm., 178: 129-136, 1999.
-
Daumas-Duport, C., Tucker, M. L., Kolles, J. H., Cervera, P., Beuvon, F. and Varlet, P., Oligodendrogliomas. Part II: a new grading system based on morphological and imaging criteria. J. Neuro-Oncol., 34: 61-78, 1997.
-
Djordjevic, B. and Snuybalski, K., Genetics of human cell lines. III. Incorporation of 5-bromo and 5-iodo-deoxyuridine into the deoxyribonucleic acid on human cells and its effect on radiation sensitivity. J. Exp. Med., 112: 509-531, 1960.
-
Menei, P., Benoit, J. P., Biosdron-celle, M., Fournier, D., Mercies, P., Guy, G., Drug targeting into the central nervous system by stereotactic implantation of biodegradable microspheres. Neurosurgery, 34: 1058-1064, 1994.
-
Benoit, J. P., Marchais, H., Rolland, H. and Van de Velde, V., Biodegradable microspheres: advances in production technology. In: Benoit, S. (Ed.), Microencapsulation methods and industrial applications, Marcel Dekker, New York, pp. 35-72, 1996.
-
Bodmeir, R., Chen, H., Davidson, R. G. W. and Hardee, G. E., Microencapsulation of antimicrobial ceftiofur drugs. Pharm. Dev. Technol., 2: 323-334, 1997.
-
Shenderova, A., Burke, T. G. and Schwendeman, S. P., Stabilization of 10-hydroxycampto- thecin in poly(lactide-co-glycolide) microsphere delivery vehicles. Pharm. Res., 14: 1406-1414, 1997.
-
Gubskaya, A. V., Lihnyak, Y. V. and Blagoy, Y. P., Effect of cryogrinding on physico-chemical properties of drugs.I. Theophylline: evaluation of particle sizes and the degree of crystallinity, relation to dissolution parameters. Drug Dev. Ind. Pharm., 21: 1953-1964, 1995.
-
Annapragada, A. and Adjei, A., Numerical simulation of milling processes as an aid to process design. Int. J. Pharm., 136: 1-11, 1996.
-
Villiers, V. M. and Tiedt, L. R., An analysis of fine grinding and aggregation of poorly soluble drug powders in a vibrating mill. Pharmazie, 51: 564-567, 1996.
-
Geze, A., Verier-Julienne, M. C., et al., Development of 5-iodo-2'-deoxyuridine milling process to reduce initial burst release from PLGA microparticles. Int. J. Pharm., 178: 257-268, 1999.
-
Rojas, J., Pinto-Alphandary, H., et al, Optimization of the encapsulation and release of b-lactoglobulin entrapped poly(D,L-lactide-co-glycolide) microspheres. Int. J. Pharm., 183: 67-71, 1999.
-
Blanco-Prieto, M. J., Bewsseghir, K., et al., Importance of the test medium for the release kinetics of a somatostatin analogue from poly(D,L-lactide-co-glycolide) microspheres. Int. J. Pharm., 184: 243- 1999.
-
Ricordi, C., Tzakis, A. G., et al., Human islet isolation and allotransplantation in 22 consecutive cases. Transplantation, 53: 407-414, 1992.
-
Warnock, G. L., Knetemoan, N. M., Ryan, E. A., Rabinovitch, A., Rajotte, A. R. V., Long-term follow-up after transplantation of insulin producing pancreatic islets into patients with type I (insulin dependent) diabetes mellitus. Diabetoligia, 35: 89-95, 1992.
-
Pyzdrowski, K. L., Kendall, D. M., et al., Preserved insulin secretion and insulin independence in recipients of islets xenografts in NOD mice. N. Engl. J. Med., 327: 220-226, 1992.
-
Chang, T. M. S., Semipermeable microcapsules. Science, 146: 524-525, 1964.
-
Limand, F. and Sun, A. M., Microencapsulated islets as bioartificial endocrine pancreas. Scinece, 210: 908-910, 1980.
-
Weber, C. J., Zabinski, S., et al., The role of CD 4+ helper T cells in the destruction of microencapsulated islet xenografts in NOD mice. Transplantation, 49: 396-404, 1990.
-
Wijsman, J., Atkinson, P., et al., Histological and immunopathological analysis of recovered encapsulated allogenic islets from transplanted diabetic BB/W rats. Transplantation, 54: 588-592, 1992.
-
Chicheportiche, D. and Reach, G., In vitro kinetics of insulin release by microencapsulated rat islets: effect of the size of the microcapsule. Diabetologia, 31: 54-57, 1988.
-
Schrezenmeir, J., Gero, L., et al., The role of oxygen supply in islet transplantation. Transplant Proc., 24: 2925-2929, 1992.
-
Lum, Z. P., Krestow, M., Tai, I. T., Vacek, I. and Sun, A. M., Xenograft of rat islets into diabetic mice. An evaluation of new smaller capsules. Transplantation, 53: 1180-1183, 1992.
-
Halle, J. P., Leblond, F. A., et al., Studies on small (< 300 mm) microcapsules. II. Parameters governing the production of alginate bead by high voltage electrostatic pulses. Cell Transplant, 3: 365-372, 1994.
-
Pariseau, J. F., Leblond, F. A., et al., The rat epididymal fat pad as an implantation site for the study of microcapsule biocompatibility. Validation of the method. J. Biomed. Mater. Res., 29: 1331-1335, 1995.
-
Robitaille, R., Pariseau, J. F., et al., Studies on small (< 350 mm) alginate-poly-L-lysine microcapsules III. Biocompatibility of smaller versus standard microcapsules. J. Biomed. Mater. Res., 44: 116-120, 1999.
-
Yin, Y., Xu, M., Chen, X. and Yao, K. D., Drug release behavior of chitosan/gelatin network polymer microspheres. Chinese Sci.Bull., 41: 1266-1270, 1996.
-
Gupta, K. C. and Ravi Kumar, M. N. V., Structural changes and release characteristics of crosslinked chitosan beads in response to solution pH, J.M.S.Pure and Appl.Chem., A36: 827-841, 1999.
-
Gupta, K. C. and Ravi Kumar, M. N. V., Preparation, characterization and release profiles of pH sensitive chitosan beads, Polym.Int., 49: 141-146, 2000.
-
Gupta, K. C. and Ravi Kumar, M. N. V., Drug release behavior of beads and microgranules of chitosan, Biomaterials, 21: 1115-1119, 2000.
-
Gupta, K. C. and Ravi Kumar, M. N. V., Semi-interpenetrating polymer network beads of crosslinked chitosan-glycine for controlled release of chlorphenramine maleate, J.Appl.Polym.Sci., 76: 672-683, 2000.
-
Albertsson, A.-C. Lofgren, A., Sturesson, C. and Sjoling, M., Design of new building block in resorbable polymers. Application in controlled drug delivery microspheres. In: Ottenbrite, R. M. (Ed.), Polymeric Drugs, Drug Administration, ACS Symposium Series 545: Chapter 14, 1994.
-
Palmgren, R., Karlsson, S. and Albertsson, A.-C., Synthesis of degradable crosslinked polymers based on 1,5-dioxepan-2-one and crosslinker of bis-e-caprolactone type. J.Polym.Sci. A: Polym.Chem., 35: 1635-1649, 1997.
-
Wakiyama, N. Juni, K. and Nakano, M., Preparation and evaluation of in vitro and in vivo of polylactic acid microspheres containing dibucaine. Chem.Pharm.Bull., 30: 3719-3727, 1982.
-
Haynes, D. and Kirkpatrick, A., Ultra-long-duration local anesthesia produced by injection of lecithin-coated methoxyflurane microdroplets. Anesthesiol, 63: 490-499, 1985.
-
Langerman, L., Golomb, E. and Benita, S., Spinal anesthesia: significant prolongation of the pharmacologic effect of tetracaine with lipid solution of the agent. Anesthesiol., 74: 105-107, 1991.
-
Mashimo, T., Uchida, I., Park, M., Shibata, A., Nishimura, S., Inagaki, Y. and Yoshiya, I., Prolongation of canine epidermal anesthesia by liposome encapsulation of lidocaine. Anesth. Analg., 74: 827-834, 1992.
-
Masters, D. B. and Domb, A. J., Loposphere local anesthetic time-release for pernieural site application. Pharm. Res., 15: 1038-1045, 1998.
-
Domb, A.J., Lipospheres for controlled delivery of substances. Us Patent, 5188837 Feb., 1993.
-
Cousins, M. J. and Bridenbaugh, P. O., In: Lippincott, J. B., (Ed.), Neural blockade in clinical anesthesia and management of Pain. Philadelphia, 1988, pp. 121-128.
-
Domb, A. J. and Maniar, M., Development of a novel parenteral drug delivery system: the liposphere delivery system. In: AAPS Meeting, Washington D.C., 1991.
-
Amselem, S., Alving, C. R. and Domb, A. J. Polymeric biodegradable lipospheres as vaccine delivery systems. Polym. Adv. Technol., 3: 351-357, 1992.
-
Amsellem, S., Domb, A. J. and Alving, C. R., Lipospheres as a vaccine carrier system: effects of size, charge, and phospholipid composition. Vaccine Res., 1: 383-395, 1992.
-
McGeer, P. L., Eccler, J. C. and McGeer, E. G., Molecular neurobiology of the mammalian brain, 2nd edn., Plenum, New York, 1987.
-
Chandler, S. H., Evidence for excitatory amino acid transmission between mesencephalic nucleus of V afferents and jaw-closer motoneurons in the guinea pig. Brain Res., 477: 252-264, 1989.
-
Byrd, K. E., Stein, S. T., Sokoloff, A. J. and Shankar, K., Carniofacial alterations following electrolytic lesions of the trigeminal motor nucleus in actively growing rats. Am.J.Anat., 189: 93-110, 1990.
-
Byrd, K. E., Domb, A. J., Sokoloff, A. J. and Hamilton-Byrd, E. L., Stimulation of trigeminal motoneurons by in vivo implantation of glutamate-impregnated microspheres. Polym. Adv. Tech., 3: 337-344, 1992.
-
Byrd, K. E. and Hamilton-Byrd, E. L., Polymeric delivery of neurotransmitters and neuromodulators. In: Domb, A. J. (Ed.), Polymeric site-specific pharmacotherapy. Wiley & Sons, New York, pp. 142-155, 1994.
-
Howard, M. A.(III), Gross, A., Grady, M. S. and Langer, R. S., Intracerebral drug delievry in rats with lesion-induced memory deficts. J. Neurosurg., 71: 105-112, 1989.
-
Kubek, M. J., Rea, M. A., Hodes, Z. I. And Aprison, M. H., Quantitation and characterization of thyrotropin-releasing hormone in vagal nuclei and other regions of the medulla oblongata of the rat. J. Neurochem., 40: 1307-1313, 1983.
-
Palkorits, M., Mezey, E., Eskay, R. L. and Brownstein, M. J., Innervation of the nucleus of the solitary tract and dorsal vagal nucleus by thyrotropin releasing hormone-containing raphe neurons. Brain Res., 373: 246-251, 1986.
-
Merchenthaler, I., Csernus, V., Csontos, C., Petrusz, P. and Mess, B., New data on the immunocytochemical localization of thyrotropin-releasing hormone in the rat central nervous system. Am. J. Anat., 181: 359-376, 1988.
-
Sharif, N. A., Quantitative autoradiography of TRH receptors in discrete brain regions of different mammalian species. Ann. Ny. Acad. Sci., 553: 147-175, 1989.
-
Rekling, J. C., Excitatory effects of thyrotropin-releasing hormone (TRH) in hypoglossal motoneurons. Brain Res., 510: 175-179, 1990.
-
Byrd, K. E., Sukay, M. J., Dieterle, M. W., Yang, L., Marting, T. C., Teomim, D. and Domb, A. J., Carniofacial and TMJ effects after glutamate and TRH microspheres implantation in proximity to trigeminal motoneurons of growing rats. J. Dent. Res., 76: 1437-1542, 1997.
-
Ravi Kumar, M. N. V. and Neeraj Kumar, Polymeric controlled drug delivery systems: Perspective issues and opportunities. Drug Dev.Ind.Pharm., 27-0218R (In Press).
-
Amass, W., Amass, A. and Tighe, B., A review of biodegradable polymers: Uses, current developments in the synthesis and characterization of biodegradable polyesters, blends of biodegradable polymers and recent advances in biodegradation studies. Polym.Int., 47: 89-144, 1998.
-
Bodmeier, R. and Wang, J. Microencapsulation of drugs with aqueous colloidal polymer dispersions. J.Pharm.Sci., 82: 191-199, 1993.
-
Benita, S., Benoit, J. P., Puisieux, F. and Thies, C., Characterization of drug-loaded poly(D, L-lactide) microspheres. J.Pharm.Sci., 73: 1721-1728, 1984.
-
Lee, K. Y., Park, W. H., and Ha, W. S., Polyelectrolyte complexes of sodium alginate with chitosan or its derivatives. J.Appl.Polym.Sci., 63: 425-432, 1997. (References therein).
-
Kidchob, T., Kimura, S. and Imanshi, Y., pH responsive release from polypeptide microcapsules. J.Appl.Polym.Sci., 63: 453-458, 1997. (References therein).
-
Kratz, F., Fichtner, I., Beyer, U., Schumacher, P., Roth, T., Fiebig, H. H. and Unger, C., Antitumor activity of acid labile transferrin and albumin doxorubicin conjugates in in vitro and in vivo human tumor xenograft models. Eur.J.Cancer, 33: S175, 1997.
-
Matsumura, Y. and Maeda, H., A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins of the antitumor agent smancs. Cancer Res., 46: 6387-6392, 1986.
-
Takakura, Y., Fujita, T., Hashida, M. and Sezaki, H., Diposition characteristics of macromolecules in tumir-bearing mice. Pharm.Res., 7: 339-346, 1990.
-
Katti, D. and Krishnamurti, N., Preparation of albumin microspheres by an improved process. J.Microencapsulation, 16: 231-242, 1999.
Corresponding author: Majeti N.V. Ravi Kumar, Department of Chemistry, University of Roorkee, Roorkee-247 667, India. mnvrkumar@mailcity.com
Published by the Canadian Society for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences.
http://www.ualberta.ca/~csps
CSPS Home | JPPS Home | Search | Subscribe to JPPS |