J Pharm Pharmaceut Sci (www.ualberta.ca/~csps) 4(1):15-23, 2001
A high-performance liquid chromatographic assay for the determination of desbutylhalofantrine enantiomers in rat plasma
Dion R. Brocks1
College of Pharmacy, Western University of Health Sciences, Pomona, CA USAPresented in part at the Pharmaceutical Congress of the Americas, March 24-29, 2001, Orlando FL.
Received January 17, 2001, Revised February 26, 2001, Accepted March 7, 2001.
Abstract
.PURPOSE: To develop a stereoselective high performance liquid chromatographic assay for determination of desbutylhalofantrine (DHF) enantiomers in rat plasma.
METHODS: After protein precipitation of 100 mL of rat plasma, racemic DHF and internal standard (quinidine sulfate) were extracted into hexane in the presence of pH 8 phosphate buffer. After transfer and evaporation of the hexane, the residue was derivatized using 0.25 M (+)-di-O-acetyl-L-tartaric acid anhydride at 4° C. After 5 min the reaction was stopped by addition of methanol in water, and the tube contents were dried, reconstituted in the mobile phase, and injected into a C18 analytical column under reverse phase conditions.
RESULTS: The derivatized enantiomers were baseline resolved and free of interference from endogenous components in plasma. Standard curves were linear (r2>0.99) over the range of enantiomer concentrations from 25-1000 ng/mL. The assay was validated to concentrations as low as 25 ng/ mL, based on 100 mL of rat plasma. The nature of diastereomers formed was found to be dependent on the temperature used during the derivatization step. In a preliminary experiment in the rat, stereoselectivity in the plasma concentrations of DHF were observed, indicating stereoselectivity in the pharmacokinetics of the metabolite.
CONCLUSIONS: The assay was sensitive and appropriate for use in pharmacokinetic studies of DHF in the rat.
Introduction
Halofantrine (HF; Figure 1) is an antimalarial drug used in the treatment of chloroquine-resistant strains of P. falciparum. As part of its side effect profile, HF causes a dose-related increase in prolongation of the electrocardiac Q-Tc interval, and in some susceptible patients Torsades de Pointes, a potentially fatal cardiac arrhythmia, has been reported (1-3). HF is chiral and is administered as the racemate. A recent in vitro study has suggested that this cardiotoxicity may be preferentially attributable to (+)-HF (3). Further, it has been suggested that the cardiotoxicity observed after administration of HF is due primarily to HF itself, and not its major circulating metabolite, (± )-mono-desbutylhalofantrine (DHF; Figure 1) (3,4). Due to this finding, and because the metabolite is roughly equipotent to HF in antimalarial activity (5), it has been suggested that the metabolite could make a safer pharmacological agent than HF itself (3).
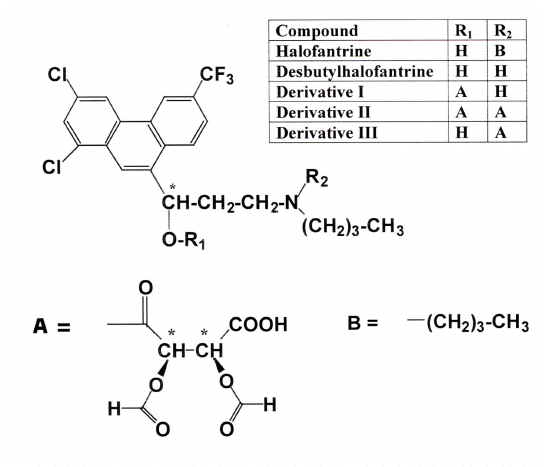
Figure 1: Structures of halofantrine, monodesbutylhalofantrine and proposed DATAAN-DHF derivatives.
The pharmacokinetics of HF are known to be stereoselective in humans and rats, with the (+) enantiomer attaining higher plasma concentrations than antipode in both species (6,7). To date there has been no attempt to determine the stereoselective pharmacokinetics of the metabolite after administration of metabolite.
We have previously reported on a stereospecific high performance liquid chromatographic (HPLC) method for HF enantiomers employing precolumn derivatization with (+)-di-O-acetyl-L-tartaric acid anhydride (DATAAN) (8,9). In this report an analytical method is presented that is capable of quantitating concentrations of DHF enantiomers in rat plasma, and which can be used to determine pharmacokinetics of DHF in that species.
Methods
Chemicals
Racemic HF HCl and DHF HCl were gifts from SmithKline Beecham Pharmaceuticals, Worthing, UK. Quinidine sulfate, the internal standard (IS), was obtained from Sigma (St. Louis, MO). Analytical grade potassium phosphate (monobasic), triethylamine, sulfuric and glacial acetic acid, ammonium hydroxide, N,N-dimethylacetamide, polyethylene glycol 400, and HPLC grade dichloromethane, acetonitrile, hexane, methanol, ethanol and 2-butanol were purchased from Fisher (Fair Lawn, NJ, USA). DATAAN, claimed purity >97%, was purchased from Fluka (Ronkonkoma, NY, USA). Water for analytical purposes was obtained using a Barnstead NanoPure Infinity water purification system (Dubuque, IA, USA).
Apparatus and chromatographic conditions
The HPLC apparatus was comprised of a Waters 710B autosampler, a Waters 600E pump/system controller (Waters, Milford, MA, USA), a Shimadzu SPD-10A UV detector, and a Shimadzu CR501 Chromatopac (Columbia, MD, USA). The chromatographic separations of DHF and its diasteromeric derivatives, and IS, were accomplished using a 100 mm ´ 8 mm I.D. Nova-Pak C18 analytical column cartridge placed in a radial compression module (Waters, Milford MA, USA). A Guard-Pak Precolumn Module containing an ODS cartridge insert was placed serially just before the analytical column.
The mobile phase for stereospecific analysis of DHF was a mixture of 53:47 [25 mM potassium phosphate:3 mM sulfuric acid:3.6 mM triethylamine]:acetonitrile containing sodium dodecyl sulfate (1.2 g/L). For the first 12 min the flow rate was set at 1.5 mL/min, after which it was increased to 2.0 mL/min. For the nonstereospecific assay of DHF used in the determination of recovery and derivatization yield, the mobile phase consisted of a 25:75 mixture of [25 mM potassium phosphate:3 mM sulfuric acid:3.6 mM triethylamine]:acetonitrile containing sodium dodecyl sulfate (1.5 g/L). This mobile phase was pumped at a flow rate of 1.5 mL/min. All mobile phases were degassed by filtration through a nylon 0.45
mm filter, and all chromatographic separations were carried out at room temperature. The UV detection wavelength was set at 254 nm.Stock solutions
A 100
mg/mL stock solution of DHF was prepared in 50% acetonitrile in water. Each day this stock solution was further diluted serially with acetonitrile in water (1:1) as needed for preparation of the standard curves. Quinidine sulfate (5 mg base/100 mL) was made up in water for use as IS. These solutions were refrigerated at 4°C in amber bottles between use. Under these conditions the analytes were stable for a period of at least 2 months, as evidenced by chromatographic analysis.For derivatizing DHF enantiomers a 0.25 M solution of DATAAN was freshly prepared in acetic acid:dichloromethane (1:4, v/v).
Sample preparation
To each 100
mL plasma sample in a 1.7 mL capped polyethylene microcentrifuge tube, 50 mL of IS solution was added. After adding 300 mL of acetonitrile, each tube was vortex mixed at high speed for 5 s, then centrifuged for 3 min at 2500 g. The protein-free supernatant was transferred to clean dry 13 x 100 mm borosilicate glass tubes containing 300 mL of Sørensen phosphate buffer (pH 8, non isotonic). After adding 4 mL of hexane, the tubes were vortex mixed for 1 min then centrifuged for 3 min at 2500 g. The organic layer was transferred to clean dry test tubes and evaporated to dryness in vacuo. To the dried residue was added 300 mL of 0.25 M DATAAN in acetic acid:dichloromethane (1:4). After incubation at 4º C for 5 min, 50 mL of methanol in water (1:1) was added and the tubes were vortex mixed for 3 s. After evaporating the tubes to dryness in vacuo, the residues were reconstituted with 120 mL of mobile phase and aliquots of 30-60 mL were injected into the HPLC.Extraction efficiency
The extraction efficiency was calculated by adding known amounts of (± )-DHF (50 and 500 ng/mL; n=4 per concentration) to 0.1 ml of blank rat plasma. The DHF was extracted into 4 mL of hexane as described above. Three mL of this hexane extract was transferred to clean tubes and evaporated to dryness in vacuo. After reconstitution of the residue in 120
mL of mobile phase, known volumes were injected into the HPLC using the nonstereospecific chromatographic conditions.The peak areas were compared to those obtained from equivalent volumes of standard solutions of DHF which were evaporated to dryness, reconstituted with mobile phase, and directly injected into the HPLC system. The determination in unextracted samples was performed in quadruplicate for each concentration.
Derivatization yield
Eight 100
mL rat plasma samples spiked with (±)-DHF (2500 ng/ml) were subjected to the protein precipitation and extraction steps outlined above. Four of the dried extracts were then derivatized, while the remaining four were set aside (underivatized). After the derivatization step was completed, 10 mL of HF solution (100 mg/mL in acetonitrile) was added to each dried residue and the tubes were reconstituted with 150 mL of the mobile phase. Equal volumes of derivatized and underivatized samples were injected into the HPLC system employing the chromatographic conditions for nonstereospecific analysis. The derivatization yield of DHF was estimated by calculating the difference in the mean underivatized DHT:HF peak height ratios between the derivatized and underivatized samples.Calibration, Accuracy and Precision
Calibration curves were constructed by adding known amounts of DHF and IS to 100
mL of blank plasma, then assaying the samples using the procedure outlined under "Sample preparation". The calibration curves consisted of concentrations of 0, 25, 50, 125, 250, 500 and 1000 ng/mL of DHF. Based on analysis of residuals, linear regression analysis was used to construct calibration curves for the peak area height ratio of analyte to IS vs. nominal analyte concentration.For the determination of intraday (within-run) and interday (between-run) accuracy and precision, on each or three separate days 5 samples of each concentration containing 25, 125 and 500 ng/mL of DHF were assayed along with independent samples used for construction of a calibration curve. Average within-run precision was determined as [
SCV% from the 3 runs]¸ 3. Between-run precision (%) was determined as 100 ´ [SD of the within-run means/average of the within-run means]. Intraday accuracy (%) was determined by 100´ [measured concentration-expected concentration]¸ expected concentration. Average intraday accuracy was calculated based on the mean of the three intraday accuracy determinations.Assessment of order of elution
We did not possess stereochemically pure, identified enantiomers of DHF enantiomers. Therefore, in order to establish the order of elution of derivatized DHF enantiomers, we used a previously developed stereospecific assay for HF and DHF in which elution order of DHF enantiomers was known (10). This assay required use of a Chiralpak AD column (Chiral Technologies, Exton, PA), with mobile phase consisting of hexane:ethanol:2-butanol:diethylamine (93:4.5:2.5:0.1) pumped at a flow rate of 0.3 mL/min, and detection wavelength set at 254 nm. A 3 h postdose plasma sample from a rat which had been given 2 mg/kg of DHF HCl was assayed using both the present assay and the method requiring a chiral column. The relative peak areas of DHF enantiomers were compared between the assays, and elution order in the presently described method was thus determined. The success of this approach required that stereoselectivity be present in the relative plasma concentrations of the enantiomers in the rat.
Test for racemization
A 50
mL aliquot of 10 mg/mL DHF solution in acetonitrile:water (1:1) was added to 100 mL of water. This sample was subjected to extraction and derivatization as outlined above. The dried residue was reconstituted in 120 uL of mobile phase and three aliquots of 30 mL were sequentially injected into the HPLC. As the derivatized DHF enantiomers eluted from the column, fractions were collected so as to separate the diastereomers. Half of the solvent was evaporated from the tubes, and then 0.5 mL of 1 M NaOH was added to each fraction to hydrolyze the diasteromers and thus liberate DHF. After letting the samples sit for 20 minutes, 4 mL of hexane were added to each fraction. The samples were vortex mixed for 45 s, centrifuged at 2500 g for 3 minutes, and the hexane layers were transferred to clean tubes. The samples were dried in vacuo, and then rederivatized as described above. The residues were reconstituted in 120 mL of mobile phase and injected into the HPLC. Evidence of racemization (appearance of antipode) was looked for in the chromatograms.Indirect characterization of derivative structure
Two sets of two tubes containing 100
mL of water and 50 mL of 100 mg/mL DHF were prepared. In each set of tubes, the enantiomers were extracted into hexane as outlined under "Sample preparation". The dried residues were derivatized with DATAAN under two conditions. In the first set of tubes, derivatization was carried out as described above (at 4° C). In the second set of tubes, 200 mL of DATAAN was added, the tubes were capped and placed in an oven for 30 min at 60ºC. After derivatization, 50 mL of methanol in water (1:1) was added to each tube and the tube contents were evaporated to dryness in vacuo. One tube of each set of tubes was set aside for injection into the HPLC using the conditions for stereospecific chromatography.To the remaining tubes, 0.5 mL of 1 M NaOH was added and the tubes were left to sit for 20 min. A 50
mL portion of the aqueous layer was reconstituted with 150 mL of mobile phase and injected into the HPLC. Hexane (2 mL) was then added to the tubes containing the remaining alkaline aqueous layers and the tubes were vortex-mixed and centrifuged at 2500 g for 1 min and 3 min, respectively. The hexane layers were then transferred to clean tubes and evaporated to dryness in vacuo. The residues was rederivatized with DATAAN at 4° C for 5 min, dried, reconstituted in mobile phase and injected into the HPLC using the conditions for stereospecific chromatography.Chromatographic analysis of both HF and DHF enantiomers
We attempted joint chromatographic determination of both HF and DHF enantiomers by extracting HF and DHF into hexane from 100
mL of rat plasma as described under "Sample preparation", above. The dried residue was then derivatized with DATAAN at 60° C for 30 min, after which 50 mL of methanol: water was added and the tube contents were dried in vacuo. The reconstituted residue was injected into the HPLC under gradient conditions. The mobile phase was 60:40 [25 mM potassium phosphate:3 mM sulfuric acid:3.6 mM triethylamine]:acetonitrile containing sodium dodecyl sulfate (1.2 g/L), run at a flow rate of 2 mL/ min. This mobile phase was switched over 30 minutes, in a linear fashion, to another mobile phase containing 47:53 of [25 mM potassium phosphate:3 mM sulfuric acid:3.6 mM triethylamine]:acetonitrile plus sodium dodecyl sulfate (1.5 g/L), and to a new flow rate of 1.2 mL/min.Pharmacokinetic evaluation
In order to test the assay for its utility in a pharmacokinetic study, two male Sprague Dawley rats were administered 2 mg/kg of DHF HCl. The drug was solubilized in a vehicle consisting of N,N-dimethylacetamide:polyethylene glycol 400 (1:11.5), to make a concentration of 5 mg/mL. The dose was injected over 1 min into a previously implanted cannula that had been surgically implanted under halothane anesthesia into the right jugular vein. Blood samples were obtained and plasma was separated for assay of DHF. The AUC from 0-8 h postdose was calculated using the log-linear trapezoidal rule.
Results and Discussion
The derivatized DHF enantiomers eluted at approximately 15 and 17.4 min after injection into the HPLC (Figure 2).
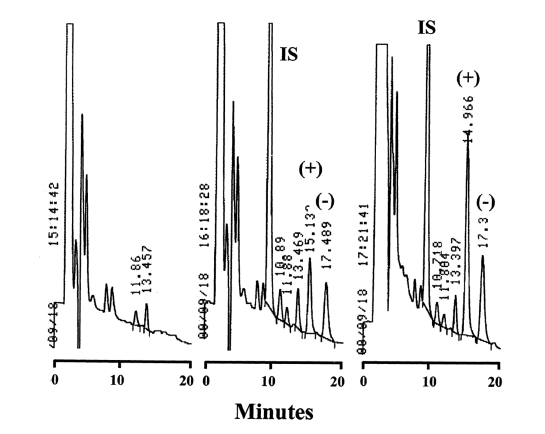
Figure 2: Chromatograms obtained from A.) Blank control plasma, B.) Sample spiked with 125 ng/mL of each desbutylhalofantrine enantiomer (denoted by (+) and (-)) and IS, and C.) plasma sample obtained 3 h after an iv dose of 2 mg/kg DHF HCl.
The IS eluted at approximately 9 min. All peaks were baseline resolved and clear of interference from endogenous components in rat plasma. The analytical run time was found to be 20 min. Stereoselectivity was present in the plasma of the rat given DHF iv (Figure 3), and along with the method using a Chiralpak AD column (10) it was determined that the first eluted diastereomer represented (+)-DHF, and the second was (-)-DHF.
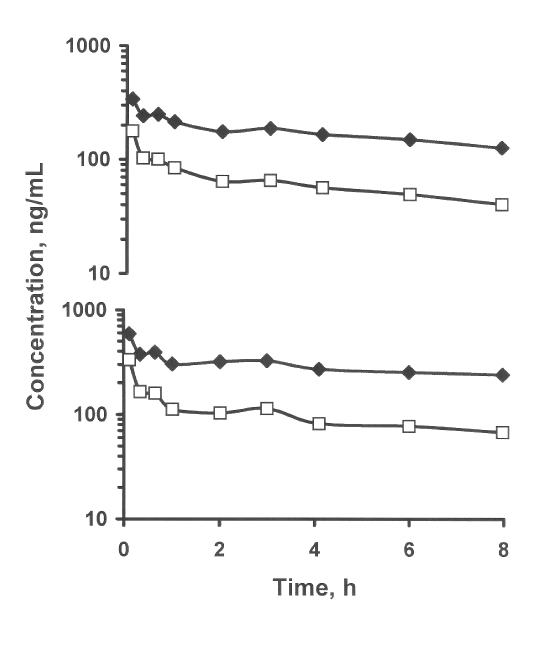
Figure 3.: Plasma desbutylhalofantrine enantiomer concentration vs. time data from two rats given an iv dose of 2 mg/kg (±)-DHF HCl. (+)-DHF is represented by closed diamonds, (-)-DHF by open squares.
Complete recovery of DHF enantiomers was achieved using the extraction method. The mean derivatization yield at a high concentration of DHF enantiomers (2500 ng/mL of each enantiomer) was over 97.5%. The assay was found to result in no observable racemization, as evidenced by a lack of antipode peak in the alkali-hydrolyzed, reextracted and rederivatized eluent fractions of each enantiomer.
The standard curves generated from the calibration samples yielded excellent linearity, with r
2>0.99. A representative standard curve could be represented by the equations (+)-DHF concentration = (peak height ratio ´ 0.000733) - 0.00520, and (-)-DHF concentration = (peak height ratio ´ 0.000570) - 0.00407. The assay was validated to 25 ng/mL based on 100 mL of rat plasma. The within and between day runs yielded RSD of <19% at 25 ng/mL, and <9% at enantiomer concentrations of 125 ng/mL and 500 ng/mL (Table 1).Table 1: Validation data of desbutylhalofantrine enantiomers in 100 mL of rat plasma, expressed as percent.

Mean accuracy ranged between 101-110%. The lower limit of quantitation, based on this analysis, was 25 ng/ mL based on 100
mL of rat plasma.Upon initial attempts to develop an assay for DHF enantiomers using precolumn derivatization with DATAAN, the same reaction conditions were adopted as used for a published HF assay (8). With that method HF was incubated in the presence of DATAAN for 30 min at 45° C, in order to allow complete derivatization of both enantiomers. At 45° C, however, inconsistent results were obtained for DHF as two pairs of peaks were observed, one pair eluting at 4 and 6 min, the other at 15 and 17 min. When the temperature was increased to 60° C for 30 min, the peaks at 15 and 17.4 min disappeared, leaving only a single pair of peaks of equal area eluting at 4 and 6 min. These peaks are visible in Figure 4 eluting at approximately 6.5 and 9 min with a more polar mobile phase and modified flow rate.
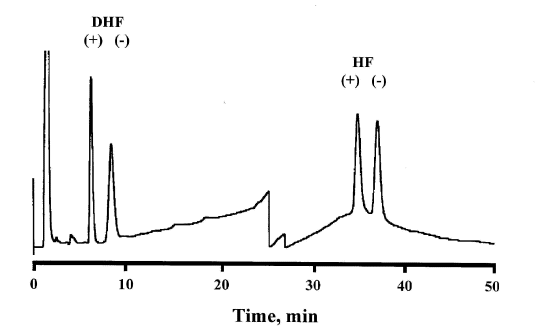
Figure 4: Chromatogram from a plasma sample containing halofantrine and desbutylhalofantrine enantiomers, dervivatized with DATAAN at 60°C for 30 min. Chromatographic conditions are described under "Chromatographic analysis of both HF and DHF enantiomers".
On the other hand, when the reaction was allowed to proceed at 4° C for 5 min, the peaks at 4 and 6 min did not appear, and we could only see peaks of equivalent area at 15 and 17.4 min. We chose to use the latter conditions for derivatization, since a.) the reaction was faster, b.) the peaks at 4 and 6 min eluted at a time where potentially interfering peaks from endogenous were co-eluting, and c.) heat was not required which could potentially lead to inadvertent racemization of the DHF enantiomers.
Theoretically, there are two sites on the DHF molecule where DATAAN could react to form diastereomers (Figure 1). One of these sites is the alcohol, in which an ester derivative could be formed, the other being the secondary amine functional group, which would result in an amide derivative. Because esters are susceptible to hydrolysis under alkaline conditions, and amides are stable, we used these properties to tentatively identify the nature of the derivatives formed. After alkali hydrolysis of the sample incubated with DATAAN at 4° C, followed by extraction into hexane and rederivatization at 4° C, large peaks eluted at 15 and 17 min. This suggested strongly that hydrolysis of ester derivative I (Figure 1) occurred after addition of alkali, thus releasing the DHF allowing reextraction into hexane and rederivatization with DATAAN. It was of note that after derivatization at 60° C, the peaks corresponding to derivative I at 15 and 17 min did not appear. Instead, two new peaks were observed eluting at 4 and 6 min. These peaks probably represented derivative II (Figure 1), which would be expected to elute earlier than derivative I in reverse phase HPLC owing to its greater polarity.
After addition of alkali to the sample derivatized at 60° C, followed by extraction into hexane and rederivatization at 4° C, the peaks corresponding to derivative I were not seen. When an aliquot of the NaOH-containing aqueous phase was directly injected onto the column at a faster flow rate of 2 mL/min, two new baseline-resolved, symmetrical peaks appeared in the chromatograms with extended retention times of 19.3 and 21.3 min. These peaks probably represented the derivative III formed as a result of cleavage of the ester DHF-DATAAN portion of derivative II. The single amide would be less polar than I, and hence would be expected to elute at a later time, as was observed. Further, derivative III would likely not be extracted into hexane under alkaline conditions due to the presence of a carboxylic acid moiety (Figure 1), which in turn would explain the lack of chromatographically discernable peaks associated with DHF after hexane extraction. Although these experiments strongly suggest these scenarios, mass spectroscopy, which was unavailable to us, would be needed for confirmation.
The ability of DHF to form different combinations of diasteromeric pairs was somewhat unexpected since under acidic conditions used in the derivatization step, the secondary amine would be expected to be protonated, and hence less reactive with DATAAN. This has been reported to be the reason why DATAAN preferentially reacts with the hydroxyl group of
b-adrenergic antagonists, which also possess secondary amine functional groups (11). The presence of heat can apparently overcome the hindrance of the protonated amine and force derivatization with DATAAN at that site on the DHF molecule.Other analytical methods are available for the stereospecific separation of HF and DHF enantiomers (10, 12,13). Each of these methods utilizes a chiral stationary phase, and one of them involves a complex scheme involving achiral chromatography before separation of the enantiomers using stereoselective chromatography (13). For our purposes we wished to develop an assay that was specific for DHF, had a short analytical run time, that could be used to assay DHF in small volumes of specimen, and which was economical. The method we have developed here has an advantage in that is has a much shorter analytical run time of 20 min vs. 40-50 min for those methods using Chiralpak AD or Chiralcel OD columns (10,12). In terms of cost, DATAAN is a relatively inexpensive reagent and a C18 column is generally much less expensive than is a chiral column, which presents an advantage to the present method. On the other hand, the method described here uses more mobile phase, which is a disadvantage. It could be argued that the current method is more sensitive than other chiral methods that have been validated to no less than 50 ng/mL of 0.5 mL of plasma. The present method, however, is disadvantageous in that it cannot simultaneously measure both HF and DHF enantiomers due to differing requirements in extraction procedure and derivatization temperatures. We opted to remove alkali and tert-butyl methyl ether from the extraction procedure developed for HF (8) because a degradation product of DHF is generated in the presence of that combination (14). Formation of the ester diastereomers is temperature dependent, and the requirement for the HF assay (>45° C) differed from that used here (4° C).
It is possible to assay a sample containing both HF and DHF enantiomers. When we jointly extracted (± )-DHF and (± )-halofantrine into hexane as described above, the resultant chromatograph yielded peaks corresponding to the derivatized HF and DHF enantiomers, which were well resolved and which had the capability of being quantified (Figure 4). The analytical run time was approximately 40 min, similar to that of the methods employing a ChiralPak AD column. The peak sizes of the HF diastereomers were similar to those of DHF. Although we did not determine extraction efficiency for HF under the conditions used for DHF extraction, this observation suggests that HF extraction into hexane is fairly high. There are some problems with the procedure, however, in that a relatively marked baseline shift occurred due to gradient elution, and the run time was long. Although HF enantiomers eluted within 40 min, an extra 15 min would be required to establish the original conditions before the next injection could be made.
The analysis of plasma samples from the rats given 2 mg/kg of (± )-DHF HCl indicated a high level of stereoselectivity in the pharmacokinetics of the metabolite (Figure 3). In the two rats studied, the plasma AUC0-8h values were 2364 and 1401 for (+)-DHF, and 813 and 506 ng·h/mL for (-)-DHF, respectively. This stereoselectivity allowed us to successfully establish the elution order in conjunction with the Chiralpak AD column. It also suggests that the metabolite shares similar pharmacokinetic properties to HF itself, which exhibits higher plasma concentration of the (+) enantiomer relative to antipode after oral and intravenous administration (6,7).
In conclusion, a novel assay was developed for DHF in rat plasma. The method is economical, had an analytical run time significantly shorter than other currently available methods, and is sensitive enough to permit use in pharmacokinetic studies involving the rat.
Acknowledgements
This work was funded by Burroughs Wellcome/AFPE (AACP-NIP grant). The author is thankful to Dr. Franco Pasutto for helpful discussion regarding the derivatization schemes, and to Mr. Tung Huynh and Mr. Shawn Nguyen for technical assistance with extraction efficiency.
References
Monlun, E., Leenhardt, A., Pillet, O., Gaston, R., Receveur, M.C., Bouabdallah, K., Longy-Boursier, M., Favarel-Garrigues, JC. and Le Bras M., Ventricular arrhythmia and halofantrine intake. Probable deleterious effect. Apropos of 3 cases. Bull Soc Pathol Exot, 86:365-367, 1993.
Nosten, F., ter Kuile, P.O., Luxemburger, C., Woodrow, C., Kyle, D.E., Chongsuphajaisiddhi, T. and White, N.J., Cardiac effects of antimalarial treatment with halofantrine. Lancet, 341:1054-1056, 1993.
Wesche, D.L., Schuster, B.G., Wang, W.X. and Woosley, R.L., Mechanism of cardiotoxicity of halofantrine. Clin Pharmacol Ther, 67:521-529, 2000.
Karbwang, J. and Na Bangchang, K., Clinical pharmacokinetics of halofantrine. Clin. Pharmacokin. 27:104-119, 1994
Karle, J.M., Olmeda, R., Gerena, L. and Milhous, W.K., Plasmodium falciparum: Role of absolute stereochemistry in the antimalarial activity of synthetic amino alcohol antimalarial drugs. Exp Parasitol, 76:345-351, 1993.
Gimenez, F., Gillotin, C., Basco, L.K., Bouchaud, O., Aubry, A.F., Wainer, I.W., Le Bras, J. and Farinotti, R., Plasma concentrations of the enantiomers of halofantrine and its main metabolite in malaria patients. Eur J Clin Pharmacol, 46:561-562, 1994.
Brocks, D.R. and Toni J.W., Pharmacokinetics of halofantrine in the rat: stereoselectivity and interspecies comparisons. Biopharm Drug Dispos. 20:165-169, 1999.
Brocks, D.R, Dennis, M.J. and Schaefer, W.H., A liquid chromatographic assay for the stereospecific quantitative analysis of halofantrine in human plasma. J Pharm Biomed Anal, 13:911-918, 1995.
Toni, J.W. and Brocks, D.R., Stereospecific high performance liquid chromatographic (HPLC) analysis of halofantrine enantiomers in rat plasma. Pharm Res, 14(suppl.):568, 1997.
Terefe, H. and Blaschke, G., Direct determination of the enantiomers of the antimalarial drug halofantrine and its active metabolite N-desbutylhalofantrine in human plasma. J Chrom B: Biomed Appl, 657:238-242, 1994.
Lindner, W. and Leitner, Ch., Liquid chromatographic separation of enantiomeric alkanolamines via diastereomeric tartaric acid monoesters. J Chromatogr, 316:605-616, 1984.
Gorichon, E., Martin, C., Na Bangchang, K., Karbwang, J., Thuillier, A., Farinotti, R. and Gimenez, F., Chiral chromatographic method to determine the enantiomers of halofantrine and its main chiral desbutyl metabolite in erythrocytes. J Chromatogr, 712:259-262, 1998.
Gimenez, F., Aubrey, A., Farinotti, R., Kirkland, K. and Wainer, I.W., The determination of the enantiomers of halofantrine and monodesbutylhalofantrine in plasma and whole blood using sequential achiral/ chiral high performance liquid chromatography. J Pharm Biomed Anal, 10:245-250, 1992.
Humberstone A.J., Currie, G., Porter, C.J.H., Scanlon, M.J. and Charman, W.N., A simplified liquid chromatographic assay for the quantitation of halofantrine and desbutylhalofantrine formed under alkaline conditions. J Pharm Biomed Appl, 13:265-272, 1995.
Corresponding Author: Dion R. Brocks, College of Pharmacy, Western University of Health Sciences, 309 East 2nd Street, College Plaza, Pomona, California, USA, 91766. dbrocks@westernu.edu
Published by the Canadian Society for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences.
http://www.ualberta.ca/~csps

