J Pharm Pharmaceut Sci (www.ualberta.ca/~csps) 5(1):12-18, 2002
Release behaviour of drugs from tamarind seed polysaccharide tablets
S. Sumathi, Alok R. Ray1
Centre for Biomedical Engineering, Indian Institute of Technology, Delhi and All India Institute of Medical Sciences, New Delhi, IndiaReceived November 22, 2000, Revised December 3, 2001, Accepted March 7, 2002
PDF version
Abstract
Purpose: This study examines the sustained release behaviour of both water-soluble (acetaminophen, caffeine, theophylline and salicylic acid) and water insoluble (indomethacin) drugs from tamarind seed polysaccharide isolated from tamarind kernel powder. It further investigates the effect of incorporation of diluents like microcrystalline cellulose and lactose on release of caffeine and partial cross-linking of the polysaccharide on release of acetaminophen. Applying exponential equation, the mechanism of release of soluble drugs was found to be anomalous. The insoluble drug showed near case II or zero order release mechanism. The rate of release was in the decreasing order of caffeine, acetaminophen, theophylline, salicylic acid and indomethacin. An increase in release kinetics of drug was observed on blending with diluents. However, the rate of release varied with type and amount of blend in the matrix. The mechanism of release due to effect of diluents was found to be anomalous. The rate of release of drug decreased on partial cross-linking and the mechanism of release was found to be super case II.
Introduction
Hydrophilic matrices are an interesting option when developing an oral sustained-release formulation. They can be used for controlled release of both water-soluble and water-insoluble drugs. The release behaviour of drugs varies with the nature of the matrix and it is the complex interaction of swelling, diffusion and erosion process (1). Release of drugs from such matrices can be controlled through their physical properties, the correct choice of gelling agent and setting up the conditions for fabrication (2).
Among hydrophilic polymers, polysaccharides are the choice material due to their nontoxicity and acceptance by regulating authorities (3). Polysaccharides like cellulose ethers (4), xanthan gum (5), scleroglucan (6), locust bean gum (7), and gaur gum (8) are some of the natural polysaccharide which have been evaluated in hydrophilic matrix for drug delivery system. Although tamarind seed polysaccharide (TSP) is used as ingredient in food material and in pharmaceuticals has not been evaluated as hydrophilic drug delivery system. TSP is a galactoxyloglucan isolated from seed kernel of Tamarindus indica. It possesses properties like high viscosity, broad pH tolerance and adhesivity (9). This led to its application as stabilizer, thickener, gelling agent and binder in food and pharmaceutical industries. In addition to these other important properties of TSP have been identified recently. They include non-carcinogenicity (10), mucoadhesivity, biocompatibility (11), high drug holding capacity (12) and high thermal stability (13). This led to its application as excipient in hydrophilic drug delivery system (11-12). Since TSP is an important excipient, the present study was undertaken to elucidate release kinetics of both water-soluble and water insoluble drugs from this matrix.
In order to predict and correlate the release behaviour of drugs from hydrophilic matrix it is necessary to fit into a suitable model. The commonly adopted model for understanding such behaviour from hydrophilic matrices is simple exponential equation (14). This model facilitates the understanding of mode of release like: whether the release is due to only diffusion or only erosion or due to both diffusion and erosion. This model has been used for this study.
Materials and Methods
Materials
Tamarind kernel powder, acetaminophen and caffeine were obtained as gift sample from Dabur India Limited. Salicylic acid from Qualigens (India), indomethacin, and theophylline anhydrous from Sigma Chemicals Company (St. Louis, U.S.A) were purchased. Microcrystalline cellulose, lactose monohydrate, magnesium stearate were purchased from Central Drug House (India). Absolute ethanol, diethyl ether, petroleum ether, glacial acetic acid, epichlorohydrin and acetone from Qualigens (India) and sodium hydroxide from E-Merck (India). All the chemicals used were of analytical grade.
Isolation of TSP
TSP was prepared following methods by Rao et al., (8,15) in three batches on a laboratory scale. To 20g of tamarind kernel powder, 200ml of cold distilled water was added and slurry was prepared. The slurry was poured into 800ml of boiling distilled water. The solution was boiled for 20 minutes under stirring condition in a water bath. The resulting thin clear solution was kept overnight so that most of the proteins and fibers settled out. The solution was then centrifuged at 5000 rpm for 20 minutes. The supernatant was separated and poured into twice the volume of absolute ethanol by continuous stirring. The product was pressed between felt. The precipitate was washed with absolute ethanol, diethyl ether and petroleum ether and then dried at 50-60°C under vacuum. The dried material was ground and sieved to obtain granules of different particle size range. The particle size range of 150-75 microns was used for preparation of tablets.
Characterization of TSP by C13 NMR and X-ray diffraction.
N.M.R. Spectroscopy:
The 13C n.m.r spectrum was recorded for TSP solution in D2O. The sample was dissolved by heating.X-ray diffraction:
Diffraction pattern of powdered TSP sample was recorded with an X-ray diffractometer (Philips X-ray generator, Holland). X-ray diffraction was performed at room temperature (30°C) with a diffractometer; target, Cu (λ=1.54Å), filter, Ni; Voltage, 40kV; current 30mA; time constant 10mm/s; scanning rate 2°/min; measured from 10-35° at full scale 200.Cross-linking of TSP
TSP was partially cross linked with epichlorohydrin (16). TSP 10g (soaked in water) and sodium hydroxide (50ml, 1 N, 54°C) were mixed with a glass rod. After homogenization (15 min), 0.5ml epichlorohydrin (6g/100g of TSP) was slowly added with continuous homogenization (15 min). The gel was then neutralized with acetic acid and washed 3 times through a sintered glass filter with a solution of water/acetone (60:40 v/v). In the final step, the resulting solid gel was washed with pure acetone over a filter. The polymer was air dried at room temperature for 72 hours and stored in airtight container. After granulation, granular fractions between 75 and 250 microns were used for preparation of tablets. Cross-linked polysaccharide was prepared in three batches.
Preparation of tablet
The total weight of the tablets (with out magnesium stearate) were 250 mg for drug: polymer ratio of 1:4 and 300 mg for drug: polymer ratio of 1:2. The ingredients (Table 1) were mixed in mixer for 5 minutes before and 5 minutes after addition of magnesium stearate (lubricant). The tablets were prepared using single-punch hand operated tablet machine (Perkin Elmer) fitted with flat-faced punches at 5 Tons compression pressure for 30 seconds. The diameter of the tablet was 13 mm and was kept constant through out the experiment.
Table 1: Formulations of various Tamarind seed polysaccharide matrices
Ingredients
Drug type (mg/tablet)
Cross linker (mg/tablet)
Diluents (mg/tablet)
Drug substancea
50
50/100
50
TSPb
200
0
180/160/140/120/100/80
Cross-linked TSP
0
200
0
Lactose/MCc
0
0
20/40/60/80/100/120
Magnesium Stearate
2.5
2.5/3
2.5
aCaffeine/Acetaminophen/Theophylline/Salicylic acid/Indomethacin,
bTamarind Seed Polysaccharide (TSP),
cMicrocrystalline Cellulose (MC)
Equilibrium swelling study
Equilibrium swelling volume (16) of partially cross-linked TSP powder and tablet were measured in water at 37°C. The drug free tablets of 250mg each (or 250 mg of powder) were placed in a 25 ml graduated cylinder to which 10 ml of water was added. After 48 hours, the equilibrium swelling volume was read directly as the volume of the gel bed. The swelling was expressed as swollen volume per unit weight of initial dry material (ml/g).
In vitro drug release study
Single face release experiments were performed at 37°C. The sample holder was immersed in 900ml-distilled water for caffeine, acetaminophen, theophylline, salicylic acid and phosphate buffer pH 7.2 for indomethacin. Sink condition was followed for the whole experiment, as the volume of dissolution medium was above 10 times the solubility of drugs in dissolution medium. Agitation of 100 rpm was provided and concentration of drug in the dissolution medium was measured as a function of time. The concentration of caffeine, acetaminophen, theophylline, salicylic acid and indomethacin were determined by monitoring the UV absorbance of the dissolution medium at 273, 242, 271, 297 and 318nm respectively (Table 2). The experiments were done for each batches and average values are reported.
Table 2: List of model drugs used for preparation of matrix tablet
Drug type
Solubility in water at 37°C (mg/ml)
Detection wave length (nm)
Caffeine anhydrous
37.0
273
Acetoaminophen
18.9
242
Theophylline anhydrous
9.9
271
Salicylic acid
3.1
297
Indomethacina
0.9
318
aPhosphate buffer pH7.2
Model used for analysis of drug release kinetics
The dissolution data were fitted according to the well-known exponential equation (14), which is often used to describe the drug release behaviour from polymeric systems.
Mt/M¥=ktn(1)
Where Mt/M¥ is the fractional release of the drug, 't' is the release time, 'k' is a constant incorporating structural and geometric characteristic of the release device (tablets) and n is the release exponent indicative of the mechanism of release. Table 3, shows an analysis of diffusional release mechanism obtained by varying the n values (17). The n values used for analysis of the drug release mechanism from the tablets were determined from log (Mt/M¥) vs. log (t) plots.
Table 3: Variation of n values with mechanism of diffusion
n
Mechanism
dMt/dt dependence
0.5
Fickian diffusion
t-0.5
0.5<n>1.0
Anomalous diffusion
tn-1
1.0
Case II transport
Zero order
n>1.0
Super case II transport
tn-1
Results and Discussion
Characterization of TSP
13C N.M.R: The 13C N.M.R spectrum of TSP is shown in Fig 1. The spectrum shows C-1 signals at 105.4, 103.4 and 100.0 ppm that are assigned to galactose, glucose and xylose residues respectively. The result complies with the reported values (18).
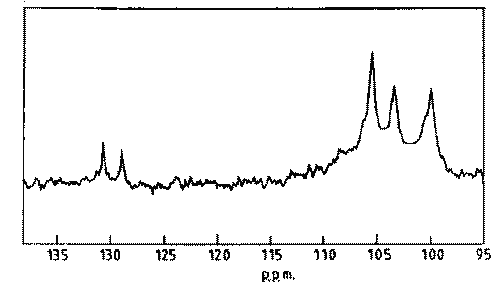
Figure 1: 13C-N.M.R. spectrum of tamarind seed polysaccharide
X-ray diffraction analysis: The X-ray diffraction pattern (Fig 2) of TSP did not show any characteristic peak, which indicates that the structure is completely amorphous.
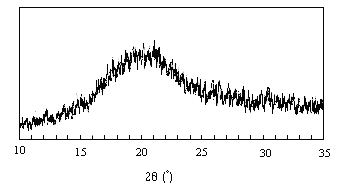
Figure 2: X-ray diffraction pattern of tamarind seed polysaccharide
The result confers with the X-ray diffraction study of tamarind xyloglucan (19). The results show that the isolated polysaccharide has similar behaviour with that reported by others. Thus polysaccharide isolated can be used in the following study.
Effect of solubility of drug
Figure 3 shows the release of drugs from TSP matrices.
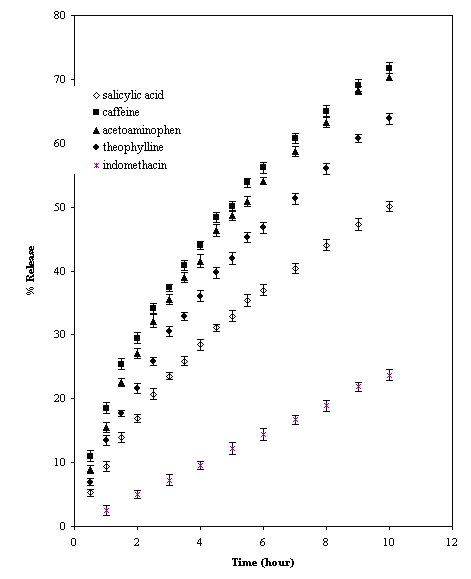
Figure 3: Release profile of drugs of different solubility from TSP tablets (mean ± SD; n = 3)
The release of drug depends not only on the nature of the matrix but it also depends upon the solubility of the drug. So the release of drugs with different solubility parameters such as caffeine, acetaminophen, theophylline, salicylic acid and indomethacin were studied (Table2). The intrinsic dissolution of the drugs in dissolution medium was determined. The procedure followed was that reported by Tarara et al (20). The 1g of drugs in 10 ml of dissolution medium were kept on a shaker at 37°C for 42 hours. 5 ml of solutions were centrifuged at 5000 rpm for 15 minutes. Then passed the supernatant through Millipore filter. The absorbances were measured at respective absorbance value and solubility values were calculated (Table 2). The rates of release of drugs from the matrices (Fig. 3) are in decreasing order of the solubility parameters. The mechanism of release of soluble drugs is anomalous (n>0.5), while indomethacin (water insoluble drug) showed behaviour of near case II or zero order release (Table 4). This indicates that the release is controlled by both diffusion and erosion phenomena. The latter dominates the release as the solubility of drug in water decreases and vice versa (20). About 50% of total loading of drug releases in 5,5.5, 7 and 10 hours for caffeine, acetaminophen, theophylline and salicylic acid respectively. The total release percent of soluble drugs in 5 hours decreases from 50% to 32% as the solubility of drug in water decreases. The total release of indomethacin in the first five hours is about 10% of total load of the tablet. The rate of release of drugs decreases with decrease in solubility of the drugs. It is because the water dissolves the drug at the surface first, and then penetrates the matrix via pores, bringing about a gelling of the polymer. Dissolved drug is then released by diffusion through the gel and finally the release rate falls as the water reaches the center due to decreased drug concentration to less than its solubility (1,21). The solubility of indomethacin in aqueous medium (phosphate buffer) is very low. Due to the slow erosion of the matrix and low solubility the amount of drug released is also less. The value of n varies from anomalous to near zero order as the solubility of drugs decreases (Table 4).
Table 4: The n value of the formulations containing drug type with D:P ratio of 1:4 and cross linker with D:P ratio of 1:2 and 1:4.
Formulation
n
Drug type
Caffeine
0.60
Acetaminophen
0.66
Theophylline
0.71
Salicylic acid
0.73
Indomethacin
0.98
Cross- linker
(1:2)
1.24
(1:4)
1.25
Effect of diluents
Figures 4 and 5 show the effect of diluents.
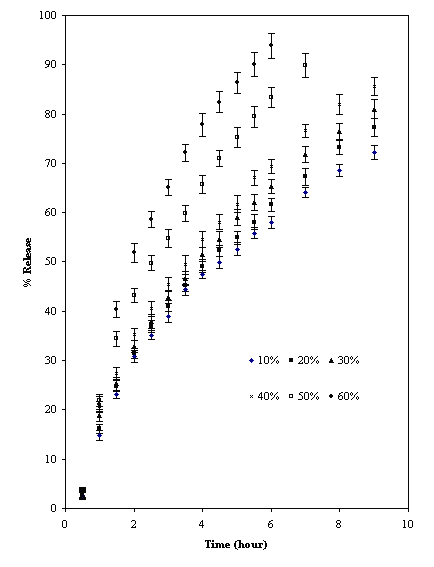
Figure 4: Effect of replacing TSP with lactose on release of caffeine (mean ± SD; n = 3)
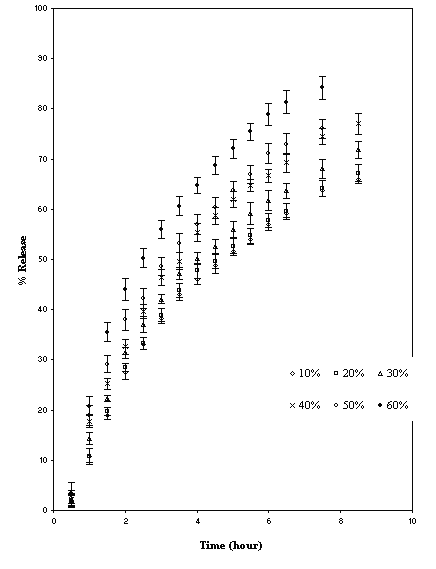
Figure 5: Effect of replacing TSP with microcrystalline cellulose on release of caffeine (mean ± SD; n=3)
Two materials were chosen for this purpose namely lactose and microcrystalline cellulose. The former is water-soluble while the latter is relatively hydrophobic. These two separately were blended with TSP and caffeine (Table 1). The mechanisms of release of caffeine from the blends were found to be anomalous (Table 5).
Table 5: The n value of formulations containing D:P ratio of 1:4 when replacing the polymer with Different amount of lactose and microcrystalline cellulose
% Replacement
n
t50
Lactose
0%
0.60
5.0
10%
0.59
4.5
20%
0.60
4.0
30%
0.61
3.75
40%
0.61
3.5
50%
0.61
2.5
60%
0.59
2.0
MC
0%
0.60
5.0
10%
0.59
4.5
20%
0.56
4.5
30%
0.56
4.0
40%
0.58
3.5
50%
0.57
3.15
60%
0.52
2.5
As the percentage of diluents increased, the kinetics of release also increased. This may be due to structural reorganization of hydrophilic polysaccharide matrix (3, 22, 23). The lactose being water-soluble would undergo dissolution and that may result in reduction in the tortuosity and or gel strength of the polymer. The T50 value of lactose and microcrystalline cellulose were nearly same up to 40% and above 40% the rate of release was faster in case of lactose (Table 5). The slow release could be due to reported interaction of tamarind seed polysaccharide with microcrystalline cellulose (19).
Effect of partial cross-linking of matrix
The partially cross-linked TSP powder and tablet had equilibrium swelling volume of 22 ml/gm and 12 ml/gm respectively. This shows that intergranular hydrogen bonds exists in the tablets due to compression like that of cross linked amylose (16). The mechanism of drug (acetaminophen) release from the two formulations of cross-linked TSP was found to be super case II (Table 3) and dissolution T50 value for drug was 8 hours (Fig.6).
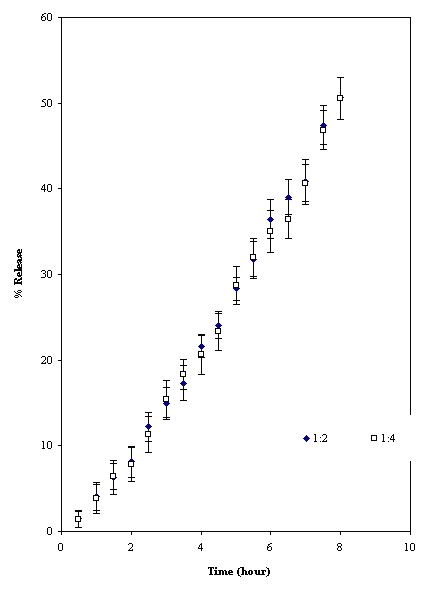
Figure 6: Release profile of acetaminophen from cross-linked TSP tablets (mean ± SD; n = 3)
The release could be sustained at constant rate for longer period than with uncross-linked material. The effect of drug loading had no effect on the percent of release (Fig. 6). The slow rate of drug release could be due to slow water penetration due to presence of numerous intergranular hydrogen bonds and presence of gel barrier (16). This shows that by controlling degree of cross-linking the release kinetics can be optimized to desire design.
Conclusion
Tamarind seed polysaccharide can be used for controlled release of both water-soluble and water insoluble types of drugs. Zero order release can be achieved taking sparingly soluble drug like indomethacin from TSP. The rate of release can be controlled by using suitable diluents like lactose and microcrystalline cellulose. For water-soluble drugs the release amount can also be controlled by partially cross linking the matrix. The extent of release can be varied by controlling degree of cross-linking.
Acknowledgements
One of the authors (S. Sumathi) is thankful to University Grants Commission, New Delhi (India) for providing financial assistance.
References
l-Carrageenan in a matrix system, I. Sensitivity to dissolution medium and comparision with Na carboxymethyl cellulose and Xanthan gum. J. Control Release, 26:119-127, 1993.
Colombo, P., Bettini, R., Massimo, G., Catellani, P.L., Santi, P. and Peppas, N. A., Drug diffusion front movement is important in drug release control from swellable matrix tablets. J. Pharm Sci, 84(8):991-997, 1995.
Vazquez, MJ., Perez-Marcos, B., Gomez-Amoza, JL., Martines., Pacheco, R., Souto, C. and Concheiro, A., Influence of technological variables on release of drugs from hydrophilic matrices. Drug Dev Ind Pharm, 18:1355-1375, 1992.
Bonferoni, M.C., Rossi, S., Tamayo, M., Pedraz, J.L., Dominguez_Gil, A. and Caramella, C., On the employment of
Ford, JL., Ribinstein, MH., McCaul, F., Hogan, JE. and Edgar, PJ., Importance of drug type, tablet shape and added diluents on drug release kinetics from hydroxypropyl methyl cellulose matrix tablets. Int J Pharm, 40:223-234, 1987.
Talukdar, M,M., and Plaizier-Vercammen J. Evaluation of xanthan gum as a hydrophilic matrix for controlled release dosage form preparations. Drug Dev Ind Pharm, 19(9):1037-1046, 1993.
Risk, S., Duru, D., Gaudy, D. and Jacob, M., Natural polymer hydrophilic matrix: influencing drug release factors Drug Dev Ind Pharm, 20(16):2563-2574, 1994.
Sujja-areevath, J., Munday, DL., Cox, PJ., and Khan, KA., Release characteristics of diclofenac sodium from encapsulated natural gum mini-matrix formulations. Int J Pharm, 139:53-62, 1996.
Khullar, P., Khar, R.K. and Agarwal, S.P., Evaluation of guar gum in the preparation of sustained-release matrix tablets. Drug Dev Ind Pharm, 24(11):1095-1099, 1998.
Rao, PS., Ghosh, TP. and Krishna, S., Extraction and purification of tamarind seed polysaccharide. J Sci Ind Research (India), 4:705, 1946.
Sano, M, Miyata, E, Tamano, S, Hagiwara, A, Ito, N. and Shirai, T., Lack of carcinogenicity of tamarind seed polysaccharide in B6C3F mice, Food and Chem Toxicol, 34: 463-467, 1996.
Burgalassi, S., Panichi, L., Saettone, MF., Jacobsen, J. and Rassing, MR., Development and in vitro/ in vivo testing of mucoadhesive buccal patches releasing benzydamine and lidocaine. Int J Pharm, 133:1-7, 1996.
Kulkarni, D, Ddwivedi, DK., Sarin, JPS. and Singh, S., Tamarind seed polyose: A potential polysaccharide for sustained release of verapamil hydrochloride as a model drug. Indian J Pharm Sci, 59(1):1-7, 1997.
Saettone, M.F., Burgalassi, S., Giannaccini, B., Boldrini, E., Bianchini, P., and Luciani, G., Ophthalmic solutions viscosified with tamarind seed polysaccharide, PCT Int. Appl. WO 97 28,787, 1997.
Korsmeyer, RW., Gurny, R., Doelkar, E., Buri, P. and Peppas, NA., Mechanisms of solute release from porous hydrophilic polymers. Int J Pharm, 15:23-25, 1983.
Rao, PS.; Srivastava, HC., Tamarind, in Whistler RL (ed), Industrial Gums. 2nd ed., Academic Press, New York, pp 369-411, 1973.
Lenaerts, V., Dumoulin, Y., and Maateescu, MA., Controlled release of theophylline from cross-linked amylose tablets. J Control Release, 15:39-46, 1991.
Langer, RS. and Peppas, NA., Present and future application of biomaterials in controlled drug delivery systems. Biomaterials, 2(10):201-213, 1981.
Gidley, MJ, Lillford, PJ. and Rowlands, DW., Structural and solution properties of tamarind-seed polysaccharide. Carbohydrate Res, 214: 299-314, 1991.
b-D-glucans to cellulose. Carbohydrate Res, 308:389-395, 1998. Mishima, T., Hisamatsu, M., York, WS., Teranishi, K.and Yamada, T., Adhesion of
Tarara, K., Yamanoto, K., and Nishihata, T., Application of model-independent and model analysis for the investigation of effect of drug solubility on its release rate from hydroxypropyl methyl cellulose sustained release tablets. Int J Pharm, 133(1-2):17-27, 1996.
Kurahashi, H., Kami, H. and Sunada, H., Influence of physicochemical properties on drug release rate from hydroxypropylmethylcellulose matrix tablets. Chem Pharm Bull, 44(4):829-832, 1996.
Lapindus, H. and Lordi, NG., Some factors affecting the release of a water-soluble drug from compressed hydrophilic matrices for oral controlled- release dosage forms. J Pharm Sci, 55:840-843, 1966.
Alderman, DA., A review of cellulose ethers in hydrophilic matrices for oral controlled-release dosage forms. Int J Pharm Tech Prod Mfr, 5(3):1-9, 1984.
Corresponding Author: Alok R. Ray, Centre for Biomedical Engineering, Indian Institute of Technology, Delhi and All India Institute of Medical Sciences, New Delhi, India. alokray@cbme.iitd.ernet.in.
Published by the Canadian Society for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences.
http://www.ualberta.ca/~csps
