J Pharm Pharmaceut Sci (www.ualberta.ca/~csps) 5(3):220-225, 2002
Studies in topical application of niosomally entrapped Nimesulide.
Aliasgar Shahiwala, Ambikanandan Misra1
Pharmacy Department, Faculty of Technology and Engineering, Maharaja Sayajirao University of Baroda, Kalabhavan, Vadodara, Gujarat, IndiaReceived 16 April 2002, Revised 11 July 2002, Accepted 26, August 2002
PDF version
Abstract
PURPOSE: A niosome based transdermal drug delivery system of Nimesulide (NIM) was developed and extensively characterized and evaluated for in-vitro performance followed by in-vivo evaluation in rats by carrageenan induced rat paw edema method. METHOD: Niosomes were prepared by lipid film hydration technique using tweens and spans. Preparation of niosomes was optimized for highest percent drug entrapment (PDE). The prepared niosomes were incorporated into 1 percent carbopol gel base and the system was evaluated for drug diffusion across human cadaver skin (HCS) using modified validated diffusion cell. The drug retention studies in niosomes were performed at refrigerated temperature (2°C - 8°C) and at room temperature (25°C±2°C) for the period of 2 months. In-vivo performance of plain drug gel, niosomally-entrapped drug in carbopol gel base and marketed formulation were evaluated using acute rat paw edema method. RESULTS: Highest mean percentage edema inhibition (PEI) was observed for niosomal nimesulide gel after 24 hours i.e. 66.68 percent ± 5.19 percent compared to plain drug gel i.e. 12.57 percent ± 1.78 percent and marketed NIM formulation i.e. 20.49 percent ± 0.91 percent. CONCLUSION: Findings of this investigation conclusively demonstrate prolongation of drug release and increase in amount of drug retention into the skin and permeation across the skin after niosomal encapsulation of NIM. Developed nimesulide niosomal gel formulation has also demonstrated enhanced anti-inflammatory activity compared to plain drug gel and marketed formulation.
Introduction
To pursue optimal drug action, functional molecules could be transported by a carrier to the site of action and released to perform their task (1). Among different carriers liposomes and niosomes are well documented for transdermal drug delivery (2-6). Niosomes are unilamellar or multilamellar vesicles where in an aqueous solution is enclosed in highly ordered bilayer made up of nonionic surfactants with or without cholesterol (chol) and dicetyl phosphate and exhibit a behavior similar to liposomes in-vivo (7). Niosomes are supposed to give desirable interactions with human skin when applied in topical preparations by improving especially the horny layer characteristics, both by reducing trans-epidermal water loss and by increasing smoothness via replenishing lost skin lipids (3). Niosomes loaded with drugs for dermal application are aimed to preferentially show interactions with the inflamed (epidermal) tissue without exerting an immediate or strong systemic action (3). It has been reported that the topical application of NSAIDs (Non steroidal anti-inflammatory drugs) over an inflamed joint results in synovial fluid drug concentrations, which exceed plasma concentrations suggesting a direct penetration of the drug into the joint (8). Nimesulide is a newer NSAID, which provides better activity profile, greater safety and higher therapeutic index (9) is selected as a drug candidate. Nimesulide is a relatively weak inhibitor of prostaglandin synthesis in vitro and appears to exerts its effects through a variety of mechanisms including free radical scavenging, effects on histamine release, the neutrophil myeloperoxidase pathway, bradykinin activity, tumour necrosis factor-α release, cartilage degradation, metalloprotease synthesis, phosphodiesterase type IV inhibition, platelet aggregation and synthesis of platelet activity factor (10-12). Object of present investigation was to encapsulate nimesulide in niosomes and incorporate the prepared niosomes into suitable dermal base to improve the therapeutic index and patient compliance of the topical dosage form.
Experimental
Materials
Nimesulide was obtained as a gift sample from Alembic Chemical Works (Baroda, India), and Spans and Tweens were purchased from National Chemicals (India) and Loba chemi (India). Cholesterol was purchased from S.D. Fine Chem. Ltd. (India). All the ingredients were used without further purification.
pH 7.4 Phosphate Buffer Saline (PBS) was prepared as described in the Indian Pharmacopoeia (1996). All other chemicals used were of analytical grade unless otherwise specified. Nimulid R Transgel, Batch no 601529, Mfg. by Panacea Biotec Ltd., New Delhi, India was used as marketed formulation.
Preparation and characterization of niosomes
Niosomes were prepared by using the lipid film hydration technique (10). Drug, Non ionic surfactant and Cholesterol were weighed as indicated in Table 1 and dissolved in chloroform/methanol system (2:1) in a 100 ml round bottom flask.
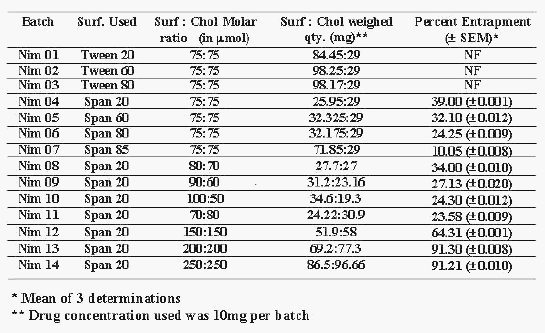
Table 1: Composition of the Niosomal batches of Nimesulide.
The solvent mixture was evaporated in a rotary flash evaporator under a vacuum of 20 inches of Hg at a temperature of 25 ± 2°C and the flask rotated at 100 rpm until a smooth, dry lipid film was obtained. The film was hydrated with 2-3 ml of PBS pH 7.4 for 45 min at room temperature (25 ± 2°C) with gentle shaking. The niosomal suspension was further hydrated at 2-8°C for 24 h. MLVs were then separated by centrifugation at 2750 rpm for 30 min. Size analysis of the niosomes were performed using ORE microscope (200 X; Russia make) and of particle size distribution of 1.5 to 3.0mm.
The formulation technique was optimized for hydration medium volume and hydration time so as to get maximum drug entrapment. All the batches were prepared three times on different days to check the reproducibility.
The prepared niosomes were analyzed for percent drug entrapment by spectrophotometric method (11) after separation of free drug. The composition of niosomes and percent drug entrapped are recorded in Tables 1 and 2. All the batches were prepared three times on different days to check the reproducibility.
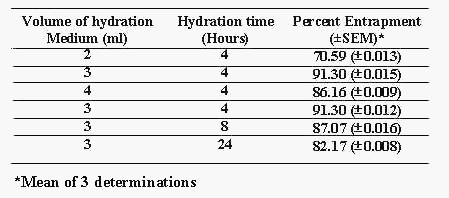
Table 2: Optimization of process variables for batch NIM 13.
Preparation of carbopol gels: Required quantity of Carbopol 934 (1%w/w) was weighed and dispersed in small quantity of distilled water to prepare an aqueous dispersion. The dispersion was allowed to hydrate for 4-5 hours. Propylene Glycol (10%w/w) and Glycerin (30%w/w) were added subsequently to the aqueous dispersion. 0.5%w/w of drug was added and properly dispersed. The dispersion was neutralized with 1%w/v NaOH solution and the pH was adjusted to 6. The final weight of the gel was adjusted to 100 g with distilled water. The entrapped air bubbles were removed using vacuum and leaving the gels overnight (Batch C1).
Niosomal gels were prepared using the same formula. For this purpose equivalent amount of niosomal suspension containing 0.5g drug was centrifuged and the pellets obtained was incorporated instead of drug (Batch C2).
Drug Retention Studies
The niosomal batch, NIM 13, and its niosomal gel formulation, Batch C2, were sealed in 20 ml glass vials and stored at refrigeration temperature (2-8°C), room temperature (25 ± 2°C). Samples from each batch at each temperature were withdrawn at definite time intervals. Samples were analyzed for the free drug to determine the leakage rate. The results are shown in Table 3.
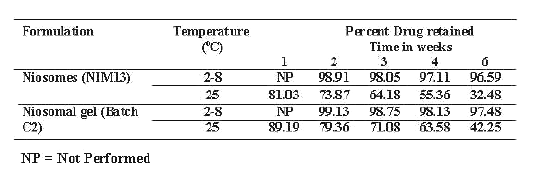
Table 3: Drug retention studies of Niosomes (NIM13) and Niosomal gel (Batch C2) at different temperature conditions.
In-vitro permeation studies
Preparation of membrane for in-vitro studies
Human cadaver skin (HCS) was obtained from ventral part of forearm of 35 years old male corpse and was stored at - 4°C. Full thickness HCS membrane was prepared by shaving the skin, punching out a tissue of approximately 2 cm 2 area, trimming away the excess fat and slicing to 500 mm thickness using a Davis dermatome 7. These slices were hydrated in pH 7.4 PBS for 24 hrs prior to use.
A vertical type of diffusion cell was designed and validated using benzoic acid disc method (12) and was use to carried out in-vitro diffusion studies. Gel containing 5 mg of drug was applied on 2.00 cm 2 area of epidermal surface of HCS tied to one end of glass permeation tube. The volume of the receptor compartment was kept to 20ml. The tube was lowered in the receptor compartment in such a way that, the dermal surface was just flush to the surface of the permeation fluid (pH 7.4 PBS, 20ml) maintained at 37.5° ± 0.5°C and stirred magnetically at 50 rpm. Aliquots were withdrawn, and replaced with the same volume of fresh buffer, at each sampling time points and analyzed for the drug content after suitable dilutions by spectrophotometric method (11). The cumulative per cent drug diffused across the HCS was calculated at each sampling point (Table 4). The mean steady state flux value along with diffusion coefficient for each formulation was calculated and recorded in table 5.
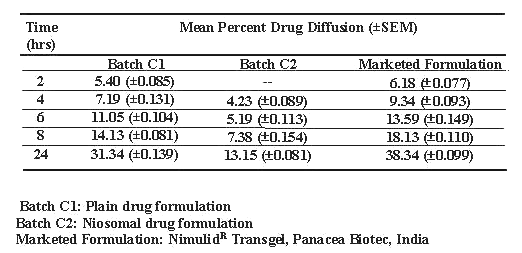
Table 4: Diffusion of drug across human cadaver skin.
Table 5: Flux and diffusion coefficient values of diffusion studies across human cadaver skin.
Drug content in the skin was calculated by subtracting the values of drug content in washing medium and in diffusion medium from the initial drug content of the formulation applied and results are recorded in table 6. The diffusion data were subjected to Student's t-test and differences greater at P < 0.005 were considered significant. All the determinations were carried out in triplicates.
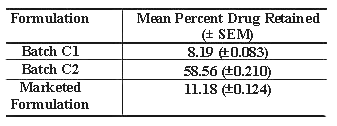
Table 6: Percent drug retained in skin after 24 hours.
In-vivo studies
The carrageenan induced rat paw edema method (13) was used as a tool to compare the efficacy of the niosomal formulation, with the plain drug formulation and marketed formulation.
Selection of Animals
White albino rats of either sex weighing between 190-230 g were selected for study. The animals were divided into four groups, each consisting 6 rats. First three groups were treated with Marketed formulation (Nimulid R Transgel, Panacea Biotec, India), test 1 preparation (batch C1, plain drug gel) and test 2 preparation (Batch C2, niosomal drug gel) respectively and the fourth group was kept as control and was treated with plain gel base with out drug to account for the effect of vehicle. Study was conducted by complete cross over design.
Methodology
Gel containing 1mg of drug was applied to left hind paw of the rats. The area of application was occluded with bandages and it was left in place for 2 hrs. The dressing was then removed and the gel remaining on the surface was wiped off with cotton. The animals were then injected with 0.1ml of 1% carrageenan solution in saline in plantar region of left hind paw and the paw volume was measured after 1 hr, 2 hr, 3 hr, 4 hr, 6 hr, 8 hr and 24 hr using water plethysmometer. The right hind paw served as a reference non inflamed paw for comparison.
Data calculation: The percentage difference between right and left paw volumes was taken as % edema produced (13). The % edema produced with test samples were subtracted from % edema produced in control group to obtain % edema inhibition by the respective group.
Results
Out of many reported (14) techniques for preparation of niosomes, lipid film hydration technique was selected to prepare niosomes containing nimesulide as reported by Azmin et al (10) with slight modification. Many simple methods namely ether injection (14), hand shaking method and sonication (14) were initially tried, but none of these methods gave satisfactory niosomal formulations. These methods resulted in extensive aggregated niosomes, poor drug entrapment or improper vesicle formation, or combination of these phenomenons. Experiments were designed to incorporate nimesulide into niosomes using lipid film hydration technique by changing the HLB (Hydrophilic Lipophilic Balance) using Tweens and Spans, keeping chol concentration same (Table 1). Prepared niosomes were analyzed for percent drug entrapment (PDE) and results are recorded in Table 1. Niosomes were not formed between HLB 17 to 14 (Tween 20, 60 and 80)(Batch No. Nim01, Nim02, Nim03) and highest entrapment (39.0% ± 0.001%) was observed at HLB 8.6 (Span 80) (Batch No. Nim04) The percent drug entrapment decreased (10.05% ± 0.008%) with decrease in HLB from 8.6 to 1.8 (Span 85)(Batch No. Nim07). When Surf:Chol ratio was changed without changing its concentration compared to drug, not much improvement in PDE was observed (23.58% ± 0.009% to 34.0% ± 0.010%) (Batch No. Nim11 to Nim08). Hence it was thought worthwhile to increase the concentration of Surf:Chol compared to drug to enhance PDE and batches were prepared. When drug to Surf:Chol ratio (75:75 mm) was raised to 2 times to 2.66 times and to 3.33 times the PDE raised from 64.31% ± 0.001% (Batch No. Nim12) to 91.30% ± 0.008% (Batch No. Nim13) to 91.21% ± 0.010% (Batch No. Nim14). Effects of process variables such as volume of hydration medium (PBS pH 7.4) and time of hydration on product were also studied (Table 2). It was observed that 3 ml of hydration medium and 4 hrs of hydration time were found to be optimum for maximum PDE i.e. 91.30% ± 0.008% (Table 2). Hence, batch Nim13 having Surf:Chol molar ratio of 200:200mm and 3 ml of hydration medium with 4 hrs of hydration time with maximum PDE (i.e. 91.3% ± 0.008%) was taken for further evaluation.
The percent drug retention studies in niosomal suspension (Nim13) and Nim13 incorporated in gel base (Batch C2) were carried out at 2-8°C (refrigerated condition) and 25°C±2°C (room temperature and the results of percent drug retained are recorded Table 3. The results indicate that drug retained above 90% in niosomes for the period of 8 weeks both in niosomal suspension and niosomal gel only at refrigerated condition.
In vitro diffusion studies of niosomal gel (Batch C2), plain drug gel (Batch C1) and marketed gel were carried out in diffusion cell using HCS. The results suggest Higuchi's diffusion controlled release model (20) for all the formulations (Table 4). The result of student's t-test (P<0.005) reveals that significant difference in drug release rates of plain drug gel, marketed formulation compared to that of niosomal gel. The mean flux values and diffusion coefficient were also compared These values were found to be 5 to7 times lower for niosomal gel as compared to plain drug gels (Table 5). Drug skin retention after 24 hrs of diffusion studies of different formulations were also compared and the maximum drug retention (58.19%) was noticed with niosomal gel compared with marketed formulation (11.18%) and plain drug gel (8.19%) (Table 6).
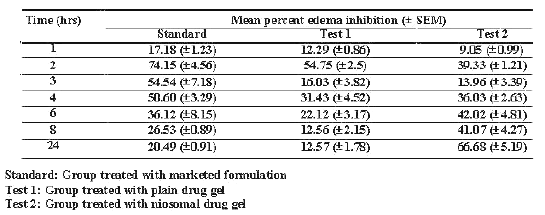
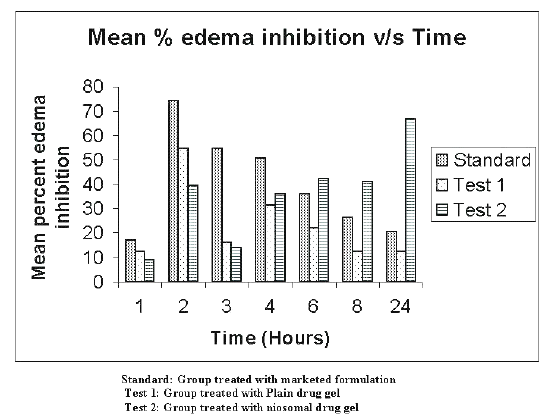
The in-vivo studies were carried out on 36 rats using carrageenan induced rat paw edema method (16). The rats were divided in to four groups each consisting of six rats. First three groups were treated with marketed formulation (Standard group), test 1 formulation (plain drug gel i.e. batch C1 - test 1 group), and test 2 formulation (niosomal drug gel i.e. batch C2 - test 2 group) respectively. The fourth group was kept as control and was treated with plain gel base without drug to account for vehicle effect. The results of the studies were calculated as percent edema inhibited and are recorded in Table 7 and shown graphically in the Figure 1.
Table 7: Percent edema inhibition by the different drug formulation.
Figure 1: Mean percent edema inhibition v/s time in hours.
The data were compared by ANOVA technique and differences greater at p< 0.005 were considered significant. Initially (up to 4 hrs), the standard group showed greater percent edema inhibition (C max -74.15%±4.56% at 2 hours) and then the effect reduced significantly with time up to 24 hrs. The group treated with plain drug gel (test group 1) showed maximum percent edema inhibition (C max - 54.75%±2.5% at 2 hours). Percent inhibition then gradually reduced to 12.57%±1.78% in 24 hrs. In case of Niosomal gel i.e. test group 2, percent inhibition was slow almost all test points initially. However, with time up to 24 hrs the percent inhibition gradually increased to 66.68%±5.19%.
Discussion
The hydrophilic-hydrophobic segments of non-ionic surfactants and a balance between them are of a paramount importance for niosome vesicle formation (21). Hence, the influence of different surf, surf: chol ratio, and drug to surf: chol ratio was studied on nimesulide entrapment in niosomes. It is also reported that the hydrophilic surfactants owing to high aqueous solubility on hydration do not reach a state of concentrated systems in order to allow free hydrated units to exist aggregates and coalesced to form lamellar structure (22,23). For this reason niosomes of nimesulide were not formed between HLB 14 to 17. The maximum drug entrapment was observed with span 20 (HLB 8.6). The influence of HLB value suggests that critical packing parameter (CPP) of a potential niosome system must take into account of presence of amphipathic or hydrophobic drugs (nimesulide) as both these substances are be incorporated into the vesicle membrane (18). Increase in chol concentration up to 50% resulted in increased PDE and any further increase did not show any influence in PDE. This increase in entrapment may be due to increase in the lipophilic behavior of the lipid bilayer of niosomes and crystallinity of the bilayer (19).
Volume of hydration medium and time of hydration of niosomes are critical process parameters during the preparation of niosomes. Improper selection of these parameters may result in improper hydration resulting into formation of fragile niosomes or drug leakage from niosomes. Optimum volume of hydrating medium of 3 ml and hydration time of 4hrs were observed to obtain maximum PDE.
The results of drug retention studies show higher drug leakage at higher temperature. This may be due to the higher fluidity of lipid bilayers at higher temperature resulting into higher drug leakage (19). The improved stability of niosomes after incorporation into the gel base may be due to prevention of fusion of niosomes.
Prolonged drug release from niosomal nimesulide gel compared to plain drug gel and marketed formulation reflected in in-vitro diffusion studies across human cadaver skin may be due to slower diffusion of drug into the skin. The lower mean flux and diffusion coefficient values of niosomal gel are suggestive of prolong drug release. The higher drug skin retention incase of niosomal gel may be due to, creation of reservoir effect for drug in skin due to deposition of other components of niosomes with drug into the skin and thereby increasing the drug retention capacity into the skin (24).
The findings of in-vitro studies were further confirmed by in-vivo studies. The reason for high initial PEI at 2 hrs (C max -74.15%±4.56%) and than gradual decrease in PEI up to 24 hrs (12.57%±1.78%) with marketed formulation may be due to the presence of alcohol and other penetration enhancers, responsible for faster absorption and excretion of the drug from the body. In case of plain drug gel significantly lower PEI at all the time points may be due to absence of penetration enhancers in plain drug gel. Slow and high drug deposition into the skin with niosomal gel may have resulted in gradual increase in the PEI between 2 hrs (39.33%±1.21%) to 24 hrs (66.68%±5.19%). The significantly higher skin retention of the niosomal drug resulting in higher partitioning of the drug in to the synovial fluid may be responsible for prolonged and enhanced anti-inflammatory activity. This is in consistent with our findings of in-vitro studies.
Findings of this investigation suggest that niosomal formulation can provide consistent and prolonged anti-inflammatory effect and may help in improving therapeutic index of the formulation and is also expected to minimize the side effects due to selective built up of drug concentrations at the site of action. However, the role of the formulation developed under this investigation can only be settled with clinical investigations on humans with emphasis on therapeutic index and side effects followed by pilot scale studies of manufacturing the product for commercial use.
Acknowledgement
The authors acknowledge University Grants Commission, New Delhi for the financial assistance and the Alembic Chemicals Works Ltd., Baroda, India for the gift sample of Nimesulide.
References
Majeti, N.V., Ravi kumar, Niraj kumar, Polymeric Controlled Drug-delivery Systems: Perspective Issues and Opportunities. Drug Dev. Ind. Pharm., 27:1-30, 2001
Singh, R. and Vyas, S.P., Selective drug delivery through and within kin using liposomes. Indian J. Pharm Sci., 58(1):9-17,1996.
Junginger, H.E., Hofland, H.E.J. and Bouwstra, J.A., Liposomes and niosomes interactions with human skin. Cosmet. Toil.,106:45-50, 1991.
Reddy, D.N., Udupa, N., Formulation and evaluation of oral and transdermal preparations of flurbiprofen and piroxicam incorporated with different carriers. Drug Dev. Ind. Pharm.,19: 843-852, 1993.
Hofland, H.E.J., Vandergeest, R., Bodde, H.E., Junginger, H.E., Bouwstra, J.A., Estradiol permeation from nonionic surfactant vesicles through human stratum corneum in-vitro. Pharm. Res., 11:659-664, 1994.
Schier, H., Bouwstra, J., Liposomes and niosomes as topical drug carriers-dermal and transdermal drug delivery. J. Control Rel., 30:1-15, 1994.
Namdeo, A. and Jain, N.K., Niosomes as drug carriers. Indian J. Pharm. Sci., 58(2):41-46, 1996.
Riess, W., Schmid, K., Botta, L., Kobayashi, K., Tomas, M. et al., Percutaneous absorption of Diclofenac. Arzneim. Forsch., 36:1092-1096, 1986.
Davis, R., Brogden, R.N., Nimesulide. An update of its pharmacodynamic and pharmacokinetic properties and therapeutic efficacy.Drugs, 48(3):431-454,1994.
Ferreira, S.H., The role of interleukins and nitric oxide in the mediation of inflammatory pain and its control by periferal analgesics. Drugs, 46(1): 1-9, 1993.
Tognella, S., Nimesulide: new clinical opportunities. Drugs, 46(1): 275-6,1993.
Verhoeven, A.J., Tool, A.T.J., Kuijpers, T.W., et al., Nimesulide inhibits platelet-activating factor synthesis in activated human neutrophils. Drugs, 46(1): 52-8, 1993.
Azmin, M.N., Florence, A.T., Handjani-Vila, R.-M., Stuart, J.E.B., Valerberghe, G., Wittaker, J.S., The effect of non-ionic surfactant vesicle (niosome) entrapment on the absorption and distribution of methotraxate in mice. J. Pharm. Pharmacol., 37:237-242,1985.
Nagoji, K.E.V., Srinivasa Rao, S., Bhanoji Rao, M.E., Kanna Rao, K.V., New spectrophotometric method for the estimation of Nimesulide in Pharmaceutical dosage formulations. East. Pharm., 42(Apr):209-210,1999.
Keshary P.R. and Chien Y.W., Mechanism of transdermal control Nitroglycerin administration. Part I- Development of finite dosing skin permeation system. Drug Dev Ind Pharm, 10:883,1984.
S.K.Kulkarni, Handbook of Experimental Pharmacology. Vallabh Prakashan, 3rd edition, pp128-131,1999.
Baillie A.J., Florence, A.T., Hume, L.R., Muirhead, G.T., Rogerson, A., The preparation and properties of niosomes non-ionic surfactant vesicles. J. Pharm. Pharmacol., 37:863-868,1985.
Israelachvili, J.N., 1985. Intermolecular and Surface Forces. Academic Press, Sydney.
Uchegdu I.F., Vyas S.P., Nonionic surfactant based vesicles (niosomes) in drug delivery. International Journal of Pharmaceutics 172:33-70,1998.
Higuchi, T., Rate of release of medicaments from ointment bases containing drugs in suspension. J.
Pharm. Sci., 50:874-875,1961.Vyas, S.P., Khar, R.K., Targeted and controlled drug delivery Novel Carrier Systems. First edition,
2002. Published by CBS Publishers and Distributors, page 253.Jousma, H., Joosten, J.G.H., and Junginger, H.E., Mesophases in mixtures of water and polyoxyethylene surfactant: Variations of repeat spacing with temperature and composition. Coll. Polyme. Sci., 266(7):640-51, 1988.
Jousma, H., Joosten, J.G.H., Gooris, G.S., and Junginger, H.E., Changes of mesophase structure of
Brij 96/water mixtures on addition of liquid paraffin. Coll. Polym. Sci., 267(4):353-64, 1989.Schreier H. et al. Liposomes and Niosomes as topical drug carriers: Dermal and Transdermal delivery. Journal of controlled release, 30:863-868, 1985.
Corresponding Author: Ambikanandan Misra, Pharmacy Department, Faculty of Technology and Engineering, M.S. University of Baroda, Kalabhavan, Vadodara-390001, Gujarat. misraan@satyam.ne.in
Published by the Canadian Society for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences.
http://www.ualberta.ca/~csps