J Pharm Pharmaceut Sci (www.ualberta.ca/~csps) 5(3):266-271, 2002
Synthesis of isatin semicarbazones as novel anticonvulsants - role of hydrogen bonding.
Surendra Nath Pandeya1, Ayyannan Senthil Raja
Department of Pharmaceutics, Institute of Technology, Banaras Hindu University, Varanasi, India.James P. Stables
Epilepsy Branch, National Institute of Neurological Disorders and Stroke, National Institute of Health, Bethesda, USA.Received 30 March 2002, Revised 25 September 2002, Accepted 9 October 2002
PDF version
Abstract
PURPOSE: A series of substituted isatin semicarbazones and related bioisosteric hydrazones were designed and synthesised to meet the structural requirements essential for anticonvulsant properties. METHODS: The structures of all synthesised compounds were confirmed by means of infrared, proton magnetic resonance spectroscopy and by elemental analyses. All compounds were evaluated for their anticonvulsant activity by maximal electroshock (MES), subcutaneous metrazol (ScMet) and subcutaneous strychnine (ScSty) induced seizure methods and their neurotoxic effects were determined by rotorod test. RESULTS: A number of isatin semicarbazones exhibited significant protection after intraperitoneal administration at the dose of 100 and 300mg/kg. Some of them showed good anticonvulsant activity in MES test in rats after per oral administration at the dose of 30mg/kg. The bioisosteric hydrazone derivatives were inactive in all tests. Compound 6-chloroisatin-3- (4-bromophenyl)-semicarbazone has emerged as the most active analogue of the series showing good activity in all the three tests and was more active than phenytoin and valproic acid. CONCLUSIONS: The results evidenced the importance of hydrogen bonding and suggested a new pharmacophore model with four binding sites essential for anticonvulsant activity.
Introduction
Epilepsy is a neurological disorder characterised by unprovoked seizures that affects at least 15 million people worldwide. Approximately 2.5 million individuals suffer from this disorder in the U.S., with 250,000 new cases diagnosed every year. Phamacotherapy is the mainstay treatment for epilepsy and the choice of antiepileptic drug for a particular patient is made according to the seizure type (1). Several new anticonvulsants (2) like oxacarbazepine, vigabatrin, lamotrigine, gabapentin, tiagabine, topiramate, felbamate, rufinamide and levetiracetam have been put into clinical practice. Despite familiarity with established antiepileptic drugs and the introduction of these new agents in the past decade, up to one third of epilepsy patients remain resistant to optimum drug treatment (3). However, the choice of these new anticonvulsants depends upon the individual factors like age, sex, type of syndrome etc. Women of childbearing age face specific problems related to the type of epilepsy and to the treatment with anticonvulsants. Also in current clinical practice combination therapy is prescribed in significant proportion of patients with epilepsy (4). Symptoms of depression were significantly more likely to appear in patients taking vigabatrin (5). These results triggered the search for newer anticonvulsants.
Recently, Dimmock (6,7) and Pandeya (8,9) have explored semicarbazones as newer chemical entities with potential anticonvulsants. The basis of the development of semicarbazones has been advocated by citing a binding site hypothesis in which there is; (a) a hydrophobic aryl ring, (b) a hydrogen bonding domain, (c) an electron donor acceptor system, and (d) an another hydrophobic aryl ring responsible for metabolism, the size of the ring can differ. To test this hypothesis some modifications were made in the structure of semicarbazones.
Isatin (indoline-2, 3-dione), a versatile heterocyclic hydrophobic molecule possessing preliminary anticonvulsant properties (10) has been selected for the hydrophobic binding site of our hypothesis.
To increase the liphophilicity of the isatin molecule, chloro substitutuents (π value 0.71) in different positions have been introduced.
(i) The hydrogen bonding domain (-CONH-) in semicarbazones has been replaced with bioisosteric non hydrogen bonding (-O-CH2 -) group.
(ii) The acetylation and benzylation of N-H group of isatin has been explored either to increase liphophilicity or to create in vivo N-H group to bind at the active site.
Lastly, the distal aryl binding site has been proposed from our earlier findings.
MATERIALS AND METHODS
Chemistry
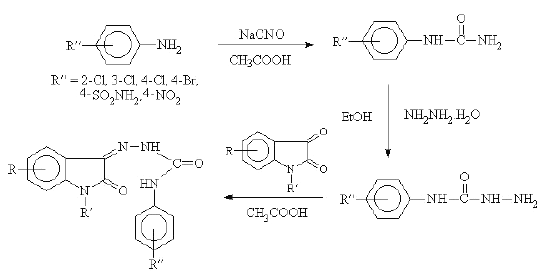
The isatin and substituted isatins (a-d) were prepared by following the procedure reported earlier (11, 12) (Scheme 1).
Scheme-1
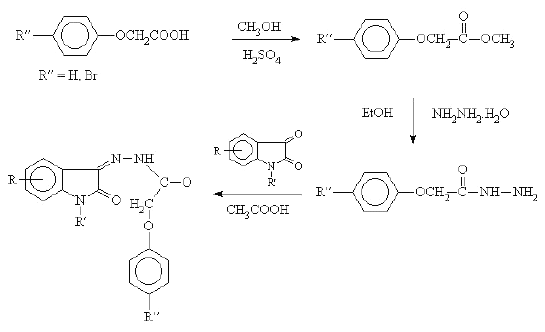
The N-acetylation of b with acetic anhydride and benzylation of a with benzyl chloride in presence of potassium carbonate and dimethyl formamide furnished N-acetyl-5-chloro isatin (e) and N-benzyl isatin (f) respectively. The substituted phenyl semicarbazides (Scheme 2) and (un) substituted phenoxy acetyl hydrazides (Scheme 3) were synthesised by using the method described in earlier literatures (13, 14). The condensation of substituted isatins a-f with aryl substituted semicarbazides and related bioisosteric hydrazides resulted in the formation of isatin semicarbazones 1-13 and 20 and isatin hydrazones 14-19, respectively.
Scheme-2
Scheme-3
Melting points were determined in open capillary tubes on a Thomas Hoover melting point apparatus and are uncorrected. The purity of the compounds was confirmed by thin layer chromatography using silica gel glass plates as stationary phase, chloroform and methanol (9:1) as mobile phase. Elemental analyses (CHN) were undertaken for all compounds and were within ± 0.4% of the calculated values. The IR spectra were recorded on a JASCO FT-IR 5300 instrument using KBr disc method and 1H NMR spectra were recorded at 90 MHz on a Jeol FX 90Q FT-NMR spectrophotometer using DMSOd6 as solvent and TMS as an internal standard.
Synthesis of isatin-3-semicarbazones (1-13 and 20)
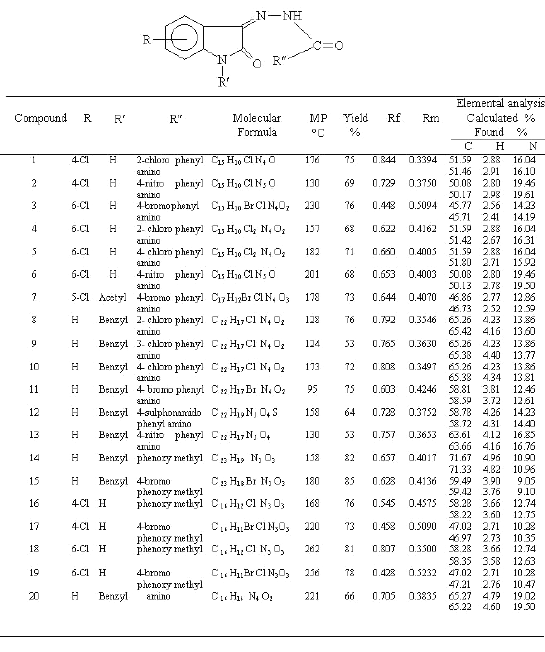
Equimolar quantities of isatin (0.003 mol) and the appropriate substituted phenyl semicarbazide (0.003 mol) were dissolved in 10 ml of ethanol (95%) containing few drops of glacial acetic acid. The mixture was refluxed for 45 minutes then cooled in ice. The resultant solid was filtered, dried and recrystallized from ethanol (95%). Percentage yield and melting points are presented in Table 1.
Table 1: Physical characterisation of synthesized compounds
1: UV (λmax, nm) 308, 242. IR (KBr ν cm-1) 3442 (secondary NH), 3306 (amide NH), 1734 (keto C=O), 1612 (C=N), 1655 (NH-CO-NH), 734 (phenyl C-H). 1HNMR (DMSOd6 d) 5.9 (s, 1H, D2O exchangeable, CO-NH), 7.7 -7.9 (m, 7H, Hetero -NH). 7: UV (λmax, nm) 303,247. IR (KBr n cm-1) 3424 (secondary NH), 3314 (amide NH), 1734 (keto C=O), 1722 (acetyl C=O), 1610 (NH-CO-NH), 1571 (C=N). 1HNMR (DMSOd6 d) 3.2 (s, 3H, N-COCH3), 5.8 (s, 1H, D2O exchangeable, CO-NH), 7.6-7.91 (m, 7H, aromatic CH), 8.7 (s, 1H, D2O exchangeable, =N-NH). 11: UV (λmax, nm) 296, 244, 207. IR (KBr n cm-1) 3490 (secondary NH), 2926 (N-CH2-), 1734 (keto C=O), 1612 (C=N), 626 (C-Cl). 1HNMR (DMSOd6 d) 3.4 (s, 2H, N-CH2-), 6.6 (s, 1H, D2O exchangeable, CO-NH), 7- 7.8 (m, 13H, aromatic CH), 8.3 (s, 1H, D2O exchangeable, =N-NH).
Synthesis of isatin-3-hydrazones 14 - 19
Equimolar quantities of isatin (0.003 mol) and parabromo or unsubstituted phenoxy acetyl hydrazide (0.003 mol) were dissolved in 10 ml of warm ethanol (95%) containing few drops of glacial acetic acid. The mixture was refluxed for 30 minutes then cooled in ice. The resultant solid was filtered, dried and recrystallized from ethanol (95%). Percentage yield and melting points are presented in table 2.
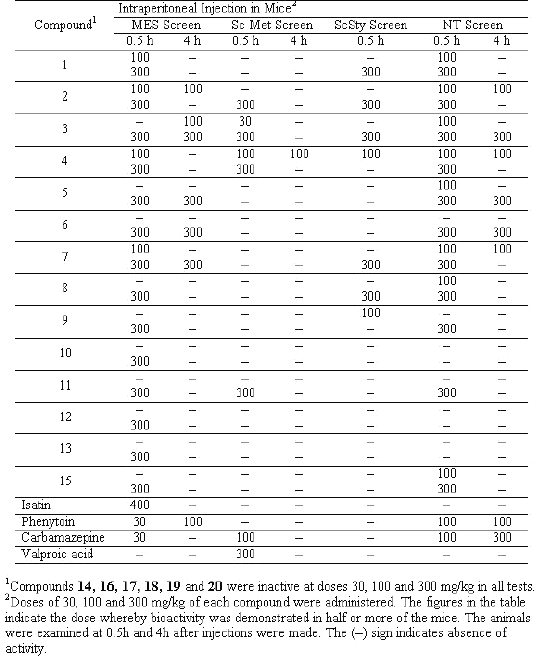
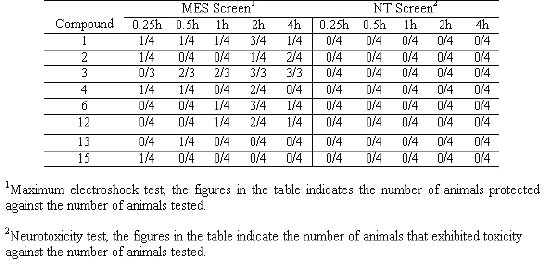
Table 2: Anticonvulsant and neurotoxicity screening of synthesized compounds
14: UV (λmax, nm) 322, 268. IR (KBr n cm-1) 3345 (secondary NH), 2926 (N-CH2-), 1734 (keto C=O), 1602 (C=N), 1248, 1190 (CH2-O), 1680 (NH-CO), 748 (aryl C-H). 1HNMR (DMSOd6 d) 3.5 (s, 2H, N-CH2-), 5.1 (s, 2H, CO-CH2), 7.1-7.2 (m, 10H, phenyl CH), 7.3-7.9 (m, 4H, isatin ring CH), 8.5 (s, 1H, D2O exchangeable, =N-NH). 17: UV (λmax, nm) 322, 268. IR (KBr n cm-1) 3419 (secondary NH), 3282 (amide NH), 2920 (-CH2-O-), 1734 (keto C=O), 1653 (C=N), 1610 (amide C=O). 1HNMR (DMSOd6 d) 5.1 (s, 1H, -O-CH2), 7.3-7.8 (m, 7H, aromatic CH), 8.6 (s, 1H, D2O exchangeable, =N-NH), 11.4 (s, 1H, D2O exchangeable, heterocyclic-NH).
Anticonvulsant screening
All the compounds were screened for anticonvulsant properties and the activity was established by following the anticonvulsant drug development (ADD) program protocol (15, 16). The compounds were administered intraperitoneally in a volume of 0.01mL/g body weight in mice at doses of 30, 100 and 300 mg/kg to 1 to 4 mice. The activities of the compounds in maximum electroshock (MES), metrazole (ScMet) and subcutaneous strychnine (ScSty) test are presented in Table 2. Further, the most active compounds were examined for oral activity in rats at dose 30 mg/kg (Table 3).
Table 3: Anticonvulsant evaluation of compounds 1, 2, 3, 4, 6, 12, 13 and 15 after oral administration (30 mg/kg) in rats
Neurotoxicity (NT) screening
The minimal motor impairment was measured in mice by the rotorod test. The mice were trained to stay on an accelerating rotorod that rotates at 10 revolutions per minute. The rod diameter was 3.2 cm. Trained animals were injected intraperitoneally with the test compounds at doses of 30, 100 and 300 mg/kg. Neurotoxicity was indicated by the inability of the animal to maintain equilibrium on the rod for at least one minute in each of the three trails. The results are shown in the Table 2.
Results
The semicarbazones were screened at 30, 100 and 300 mg/kg intraperitoneally in mice for anticonvulsant activity using the procedure described previously. In the first series of the experiments the compounds were administered by i.p. route and the MES, ScMet and ScSty tests were performed for each compound. In the initial evaluation of the anticonvulsant activity there is a clear cut differentiation in the activity of aryl substituted semicarbazones (compounds 1 to 15) showing anticonvulsant activity over those with phenoxy substituted derivatives except 15. This finding clearly demonstrates the importance of hydrogen bonding domain present in the aryl substituted semicarbazones and absent in aryloxy substituted derivatives. Among the compounds showing anticonvulsant activity from 1 to 15, compound with more lipophilic substitution like chloro group in the isatin molecule 1 to 7, are more active than those where there is no chloro substitution in the isatin ring (8 to 15).
Among the N-substituted derivatives of isatin the acetyl group has appeared to be more favourable (activity at 100 mg/kg) as compared to the benzyl substituted derivatives (activity at 300 mg/kg), which may be due to easier deacetylation as compared to debenzylation so that the NH group becomes free in vivo for hydrogen bonding with the receptor.
Some compounds (2, 3, 4 and 11) have also shown activity in both MES and ScMet screen exhibiting a broad spectrum anitconvulsant activity. Among these compound 2, 3 and 4 showed activity in all the three tests at the dose of 100 mg/kg.
Next, in the second series of experiments the most active compounds (1 to 4, 6, 12, 13 and 15) were administered by oral route at 30 mg/kg in rats in MES test and neurotoxic effects examined at 0.25, 0.5, 1, 2 and 4 h intervals. Compound 3 [6-chloroisatin-3- (4-bromophenyl) semicarbazone] exhibited excellent activity (50% protection after 0.5 h and 100% protection after 2 h at 30mg/kg) in MES test after per oral administration. Compounds 1, 4, 6 and 12 showed maximum protection after 2 h of administration and compound 2 showed maximum protection after 4 h. Only moderate activity was observed by compounds 13 and 15 after per oral administration. None of the compounds showed neurotoxicity after oral administration at 30 mg/kg in rats.
Discussions
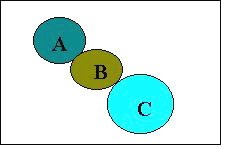
In the present series of the compounds many of them showed good anticonvulsant activity at 100 mg/kg as compared to isatin (17) (MES 400 mg/kg). So there is an increase in anticonvulsant activity by our molecular modifications. In the semicarbazone series and urelylene anticonvulsants Dimmock has proposed a binding site hypothesis for these compounds eliciting anticonvulsant activity (Figure 1).
Figure 1: Proposed binding site of urelylene anticonvulsants. The locations A and C are considered to be hydrophobic binding areas and B is hydrogen bonding site.
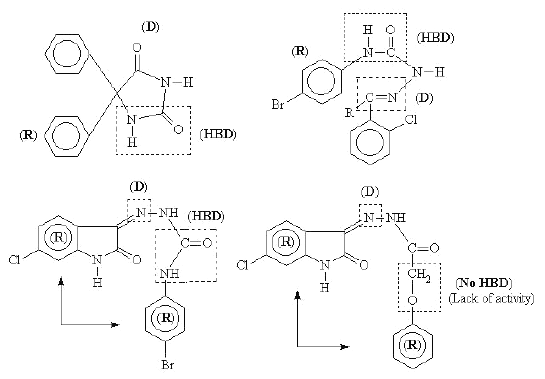
A scrutiny for certain selected structures for active anticonvulsants has been shown to possess a hydrophobic unit (R), an electron donor group (D) and hydrogen donor acceptor unit (HBD) as shown in Figure 2.
Figure 2: Structures of anticonvulsants showing the general pharmacophore model for anticonvulsant activity.
In our present series of compounds the active compound 3 possess all the requirements essential for anticonvulsant activity as proposed by Dimmock and others. The replacement of the amide bond responsible for hydrogen bonding in the phenoxy substituted derivative 16 clearly establishes the requirement of this unit for anticonvulsant activity (shown in Figure 2).
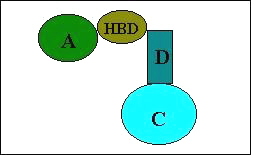
Thus our new proposal for a pharmacophore model includes not only three factors but also an additional hydrophobic binding site C shown in Figure 3 for bioactivity.
Figure 3: Suggested pharmacophore model for anticonvulsant activity. A and C are hydrophobic binding site, HBD is hydrogen-bonding site and D is electron donor group.
In conclusion these results give a new look at the pharmacophore model suggested earlier by several workers, which will account for bioactivity of majority of compounds. From our present work, compound 3 has emerged as lead molecule whose further molecular modification is under investigation.
Acknowledgements
The authors are thankful to the Head, Department of Pharmaceutics, Institute of Technology, Banaras Hindu University for providing facilities. One of the authors (A. Senthil Raja) is grateful to University Grants Commission, New Delhi for the award of Junior Research Fellowship.
References
a-(2-pyridyl)-4-quinolinemethanol. J Am Chem Soc, 2: 2695-2697, 1946.
Solomon, G.E. and Plum, F., Clinical management of seizures: A guide for the physician. W. B. Saunders Co, Philedelphia, 1976.
Sabers, A. and Gram, L., Newer anticonvulsants. Comparative review of drug interactions and adverse effects. Drugs, 60(1): 23-33, 2000.
Kwan, P. and Brodie, M. J., Early identification of refractory epilepsy. N Engl J Med, 342: 314-319, 2000.
Rall, T. W., and Schleifer, L. S., Drugs effective in the treatment of epilepsies: Goodman and Gilmans The Pharmacological Basis of Therapeutics. 6th ed., Macmillan Publishing Co, INC, New York, pp 448-474, 1980.
Johnston, S. J., Vigabatrin and behavioural disturbances. Lancet, 335: 606, 1990.
Dimmock, J.R. and Baker, G.B., Anticonvulsant activities of 4-bromobenzaldehyde semicarbazone. Epilepsia, 35 (3): 648-55, 1994.
Dimmock, J.R., Vashishtha, S.C and Stables, J.P., Urelylene anticonvulsants and related compounds. Pharmazie, 55: 490-494, 2000.
Pandeya, S.N., Yogeeswari, P. and Stables, J.P., Synthesis and anticonvulsant activity of 4-bromophenyl substituted aryl semicarbazones. Eur J Med Chem, 35: 879-886, 2000.
Pandeya, S.N., Mishra, B., Singh, P.N. and Rupainwar, D.C., Anticonvulsant activity of thioureido derivatives of acetopheonone semicarbazones. Pharmacol Res, 37(1): 17-22, 1998.
Sareen, K., Kohli, R.P., Amma M.K.P. and Gujral, M.L., Anticonvulsant drugs based on the neurochemistry of seizures. Ind J Physiol and Pharmacol, 6: 87-93, 1962.
Henry, G., Blatt, A.H., Organic Synthesis Collective Volume I. 2nd ed., John Wiley and Sons, New York, pp 327-334, 1964.
Senear, A.E., Herbert, S., Mead, J.F.and Keopfli, J.B., The synthesis of potential antimalarials. 7- chloro -
Pandeya, S.N., Mishra, V., Ponnilavarasan, I. and Stables, J.P., Anticonvulsant activity of p-chloro phenyl substituted aryl semicarbazones- the role of primary terminal amino group. Pol J Pharmacol, 52: 283-290, 2000.
Pandeya, S.N., Manjula, H. and Stables, J.P., Design of semicarbazones and their bioisosteric analogues as potential anticonvulsants. Pharmazie, 56(2): 121-124, 2001.
Krall, R.L., Penry, J.K., White, B.G., Kupferberg, H.J. and Swinyard, E.A., Antiepileptic drug development: Anticonvulsant drug screening. Epilepsia, 19: 409-428, 1978.
Porter, R.J., Hessie, B.J., Cereghino, J.J., Gladding, G.D., Kupferberg, H.J., Scoville, B. and White, B.G., Advances in the clinical development of antiepileptic drugs. Fed Proc, 44(10): 2645-2649, 1985.
Popp, F.D., Potential anticonvulsants. IX. Some isatin hydrazones and related compounds. J Heterocyclic Chem, 21: 1641-1645, 1984.
Corresponding Author: S.N. Pandeya, Department of Pharmaceutics, Institute of Technology, Banaras Hindu University, Varanasi, 221005, India. snpandeya@rediffmail.com
Published by the Canadian Society for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences.
http://www.ualberta.ca/~csps