J Pharm Pharmaceut Sci (www.ualberta.ca/~csps) 6(3):308-314, 2003
Chemotherapy induced gastrointestinal toxicity in rats: involvement of mitochondrial DNA, gastrointestinal permeability and cyclooxygenase -2.
Jaime A. Yáñez 1,2 , Xiao Wei Teng 1 , Kathryn A. Roupe 1,3 , Marc W. Fariss 1,3 , Neal M. Davies* 1,2,3,4
1 Department of Pharmaceutical Sciences, 2 Summer Undergraduate Research Fellowship Program, 3 Pharmacology and Toxicology Research Program, 4 Cancer Prevention and Research Centre, College of Pharmacy, Washington State University, Pullman, Washington, USAReceived 8 August 2003, Revised 12 September 2003, Accepted 12 September 2003
PDF version
Abstract
PURPOSE: The gastrointestinal damage induced by xenobiotics is occurring more frequently and with greater toxicological significance than previously thought. Although there are some pre-liminary clinical studies and reports, there does not appear to be an extensive examination of gastrointestinal toxicity of various chemotherapeutic agents in the rat. This study was undertaken to examine the suitability of a rat model to detect the gastrointestinal damage after administration of various anti-neoplastic agents including etoposide, teniposide, melphalan, 5-fluorouracil, methotrexate and cisplatin. METHODS: Acute toxic doses of indomethacin and chemotherapeutic agents were administered to rats. The urinary excretion of orally administered sucrose and 51Cr-EDTA were measured as markers of gastroduodenal and intestinal permeability, respectively. Cyclooxygenase-2 messenger RNA and mitochondrial DNA damage were measured as toxicological endpoints. RESULTS: Each anti-neoplastic agent examined induced appreciable and significant dose-dependent increase in gastrointestinal permeability that correlated with gross toxicological and pathological changes to the gastrointestinal tract including ulceration and bleeding. COX-2 mRNA was upregulated > 2 fold in intestinal mucosa with enteropathy and dose-dependent mitochondrial oxidative damage was apparent in gastric and intestinal mucosa. After administration of each drug, the rats presented with histological evidence of drug-induced gastroenteropathy, ulceration and increased cecal hemoglobin. CONCLUSIONS: The rat appears to be a suitable model to study gastrointestinal toxicity of chemotherapeutic agents and non-steroidal anti-inflammatory drugs. Damage to mitochondrial DNA occurs in both the gastric and intestinal epithelium after the administration of these agents and may be an important factor in the pathogenesis and resolution of gastrointestinal toxicity.
Introduction
There are a variety of classes of chemotherapeutic agents used effectively in anti-neoplastic chemotherapy. With their tremendous clinical utility, there is a large arsenal of chemotherapeutic agents available on the market, and many more currently in clinical and pre-clinical trials. The magnitude of use of these agents has demonstrated therapeutic benefits in a wide variety of cancers including gastrointestinal (GI) cancers from the esophagus to the large intestine (1-3). Paradoxically, administration of chemotherapy is well known to be associated with various side effects, with toxicity to the GI tract being a major clinical concern. GI toxicity is often a major cause of cancer treatment-related morbidity (4). Stomatitis, the mucositis of oral mucosa, is the best-characterized manifestation because it results in symptoms in an area accessible to routine examination. However, mucositis may affect any part of the GI tract. Chemotherapy induced GI toxicity is not simply confined to the upper gastroduodenal mucosa but also extends along the entire GI tract from the mouth to anus and is a common limiting factor that prevents further dose escalation (5).
The routine assessment of GI toxicity of chemotherapy is based on the patient's symptoms. The severity of stomatitis and frequency of diarrhea are the commonly used criteria for grading GI toxicity. The severity of the involvement of the oral mucosa may not reflect the extent of the mucositis in distal locations and the frequency of diarrhea may be affected by other factors associated with nutrition, and/or tumors in the GI tract. Mucositis is often painful and can limit nutritional intake and decrease a patient's willingness to continue treatment. Severe mucositis may necessitate hospitalization, enteral or parenteral nutrition, and use of narcotics (6). Muscositis increases risks of secondary infections and may even prove a nidus for systemic infections. Mucositis may necessitate interruption of chemotherapy cycles or radiotherapy, which may compromise the locoregional response. Optimal understanding and management of mucositis that results in maintenance of dose-intensity could, hence, influence morbidity, treatment efficacy, and even patient survival.
In recent years, there have been considerable advances in the understanding of the pharmacological and molecular mechanisms involved in the pathogenesis of GI toxicity particularly of non-steroidal anti-inflammatory (NSAID) induced GI damage (7-8). The early disruption of the intestinal barrier, before pronounced histological changes are evident, is of interest because it may provide pathophysiologic information concerning the early phase of the mucosal injury induced by NSAIDs and cytotoxic drugs (9). Although there is now awareness of non-prostaglandin dependent mechanisms of NSAID effects and side effects in the GI tract including inhibition of oxidative phosphorylation in mitochondria (10-11), the importance of NSAID inhibition of prostaglandin (PG) synthesis remains. It is generally believed, however, that GI toxicity is ascribed mainly to NSAID-induced inhibition of PG synthesis. The inhibition of PG synthesis causes loss of epithelial cell integrity within the GI tract, and it is responsible for the beneficial effects of NSAIDs. In addition, studies also have demonstrated that GI inflammation is associated with oxidative stress, which appears to be a critical mediator of injury after administration of various xenobiotics. Oxidative stress is of substantial clinical importance not only because oxidants are common in inflammation, but also, because they can lead to mucosal barrier hyperpermeability and, in turn, lead to the initiation and/or pertubation of mucosal inflammation and injury (11).
Interestingly, the mechanisms involved in the sequence of events leading to GI toxicity of chemotherapeutic agents as well as the temporal aspects are not fully known. However, they may share some common pathogenic mechanisms with NSAIDs. Knowledge about the primary factors initiating this injury could be of great importance in the search for protective strategies against GI toxicity due to cytotoxic therapy.
As the intestinal epithelium has a rapid cell turnover, it is vulnerable to chemotherapy. From limited clinical studies, it appears that reduction in mucosal barrier integrity is apparent with chemotherapeutic agents from a variety of classes of chemotherapy (12-16). Interestingly, the time course of these permeability changes has not been studied, which makes comparative studies between different chemotherapeutic agents difficult. In addition, the influence of circulating drug concentration has not been considered. Morphological changes in the mucosa post-chemotherapy may affect function acutely. These involve changes in the regulation on the tight junction between adjacent enterocytes. Intestinal permeability may be a morphological correlate of the early disruption of the intestinal barrier (12-16). Luminal aggressive factors such as immmunoreactive antigens and endotoxin are thus allowed access to the mucosa and can lead to the initiation or continuation of the inflammatory process and mucosal damage. Disturbance of enterocytes may also impair the ability to combat free oxygen radicals that are partly responsible for cell damage induced by chemotherapy. Mitochondrial oxidative damage to intestinal enterocytes may precede these permeability changes and be further perturbed by the inflammatory insult. This may be important for the transition from the inactive to active (flare up) phases of inflammation in which intestinal oxidants and pro-inflammatory molecules periodically create a vicious cycle that leads to sustained oxidative stress, increased permeability, inflammation, and tissue damage.
Substantial efforts have been made to develop non-invasive methods of detecting GI side effects. Methods based upon measurement of the GI permeability probes have been found to site-specifically measure gastrointestinal permeability in basic and clinical studies (9, 17-20). Most of the work carried out has been upon exposure to NSAIDs, where there is a significant increase in the urinary excretion of non-invasive probes, such as sucrose and 51Cr-EDTA, indicating increased permeability and tissue damage to the gastroduodenum and small intestine, respectively. We have previously tested and confirmed the suitability of the rat as an animal model of GI permeability using both 51Cr-EDTA and sucrose. Sucrose is a specific marker of the upper GI tract (gastroduodenum) whereas 51Cr-EDTA is used as a marker of small intestinal damage. (9) These methods are non-invasive, reproducible and, together they provide a convenient assessment of the entire GI tract (17-18). Direct measurements of GI toxicity (e.g., ulceration), throughout the GI tract correlates with increased GI permeability (21). Permeability tests are convenient, sensitive and non-invasive markers of gastrointestinal damage.
Given the apparent parallels between the occurrence of GI adverse effects in rats and humans with NSAIDs it is of interest to examine whether GI permeability and mitochondrial DNA changes can also be induced in the rat after chemotherapy. The aim of this study is to examine the suitability of the rat as a model for further investigating chemotherapy-induced GI damage using various biomarkers endpoints including GI permeability probes, mitochondrial DNA and COX-2 mRNA.
Methods and Materials
Chemicals
Indomethacin, etoposide, teniposide, sucrose, sucrose assay kit, hemoglobin assay kit, melphalan, 5-fluorouracil, methotrexate, cisplatin and methylcellulose were purchased from Sigma Chemicals (St. Louis, MO, USA). 51Cr-EDTA was purchased from NEN Life Sciences Products (Boston, MA, USA). RNeasy® Mini Kit 50 was obtained from Qiagen Inc (Stanford, CA, USA).
Animals and Surgical Procedures
Male Sprague-Dawley rats (175-195 g) were obtained from Charles River Laboratories (Wilmington, MA, USA). They were housed in individual metabolic cages with wire mesh floors allowing separate quantitative collection of urine and feces. Animals were fed a standard rat chow and allowed free access to food and water for the duration of the experiment. Ethics approval for animal experiments was obtained from Washington State University.
Drugs, 51Cr-EDTA, sucrose, dosing and assay
All drugs and placebos were in the form of a suspension (2% methylcellulose/0.5 mL water) and were administered orally to rats (n = 5/group) using an 18 gauge 5 cm curved feeding needle attached to a 1 mL syringe. Dosages of NSAIDs and chemotherapeutic agents were all based on a toxic range of doses previously demonstrated to induce gastrointestinal toxicity in animal studies. (22-32)
The effect on sucrose permeability was examined 1 hour post-dose while the effects on 51 Cr-EDTA were examined 24 hours post-dose. 51 Cr-EDTA relative permeability was determined by calculating the radioactivity present in urine samples as a percentage of the administered dose as previously described (17). Sucrose concentration was measured using a commercially available assay kit (Sigma Chemicals, St. Louis Mo. USA) utilizing a UV-VIS spectrophotometer at 340 nm and relative permeability determined by calculating the concentration (amount) in urine as a percentage of the administered dose (18). Cecal hemoglobin concentrations were measured using a commercially available kit (Sigma Chemicals, St. Louis Mo. USA) at 530 nm and concentration calculated directly from a standard curve.
Quantitative Real-Time RT-PCR
Total cellular RNA was isolated from gastric and intestinal tissue with evidence of macroscopic ulceration using the RNeasy Mini, RNA isolation kit from Qiagen (Valencia, CA, USA) per the manufacturer's protocol. Total RNA was eluted from the matrix with 35 mL of RNase-free water. Residual genomic DNA was removed by incubating the RNA solution with 15 units of RNase-free DNase I in 2 mM MgCl2 for 10 min at 37°C, followed by 5 min at 90°C to inactivate the DNase. DNase-treated RNA solution (25 mL) was reverse-transcribed to cDNA as previously described. (33) The amount of COX-2 mRNA relative to the 18s rRNA endogenous control was determined using real-time, quantitative PCR.
The PCR was performed in the Perkin-Elmer Applied Biosystems GeneAmp 5700 Sequence Detection System (Foster City, CA, USA) using the SYBR green PCR kit as recommended by the manufacturer. Briefly, the reaction medium contained 2.5 L of the 10x SYBR green buffer, 1 mM dA, dG, dC and dUTP, 2 mM MgCl2, 0.25 units of uracil N-glycosylase, 0.625 units of Amplitaq Gold DNA polymerase, 250 nM of the forward and reverse primer, (Table 1) 5 L of a 1:10 dilution of the cDNA and water to 25 L.
Table 1: Primer sequences for rt-pcr.(12)
The reactions were performed in the MicroAmp 96-well plate capped with MicroAmp optical caps (Perkin-Elmer Applied Biosystems, Foster City, CA, USA). The reactions were incubated at 50°C for 2 min to activate the uracil N'-glycosylase and then for 10 min at 95°C to inactivate the uracil N'-glycosylase and activate the Amplitaq Gold polymerase. The reactions were performed for 40 cycles of 15 sec at 95°C, 30 sec at 55°C and 30 sec at 72°C.
The data generated by real-time PCR were plotted as the Rn fluorescence signal versus the cycle number. The Applied Biosystems, Inc. (ABI) 5700 sequence detection system software from Perkin Elmer calculates the DRn using the equation DRn = (Rn+) - (Rn-), where Rn+ is the fluorescence signal of the product at any given time and Rn- is the fluorescence signal of the baseline emission during cycles 3 to 15. An arbitrary threshold was set at the midpoint of the log DR n versus cycle number plot. The C t value is defined as the cycle number at which the DR n crosses this arbitrary threshold. The amount of COX-2 cDNA relative to the 18s RNA endogenous control was determined using a modification of the 2 - D DCt method as described in the ABI user bulletin number 2. The amount of COX-2 mRNA relative to 18sRNSA was calculated equal to 2 -D DCt where DC t = C tCOX-2 - C 18srRNA (33).
Mitochondrial and Nuclear DNA
A quantitative polymerase chain reaction (QPCR) technique was developed based on the work of Van Houten et al . (34-35) Damage in mitochondrial DNA (mt DNA) can be assessed without the need for isolation of mitochondria or mitochondrial DNA. Gene specific DNA damage provides more insight into the role played by oxidative stress in disease and mitochondrial DNA damage is an excellent biomarker of oxidative stress in epithelial cells.. (Table 2)
Table 2: Primer sequences for rat qpcr.(34-35)
DNA was isolated using Qiagen® genomic tip and genomic DNA buffer set kit for mammalian DNA extractions (Valencia, CA, USA). DNA quantitation utilized the PicoGreen® dsDNA Quantitation Kit (Molecular Probes, Eugene, OR, USA). Picogreen® was used to quantify dsDNA fragment. QPCR involved the use of GeneAmp XL PCR kit (Applied Biosystems, Branchburg, NJ, USA) and dNTPs (Pharmacia, Peapack, NJ, USA). Primers were based on sequences already optimized by Van Houten. (34-35) After fluorescent readings for all samples are taken using a CytoFluor fluorescence multiwell plate reader Series 4000 (Applied Biosystems, Framingham, MMA, USA) and subtracted from the no template controls relative amplification is calculated.
Statistical analysis
Differences between two means were determined by Student unpaired t-test. Differences between more than two means were determined by one-way ANOVA followed by Duncan's multiple range test at a 0.05.
Results
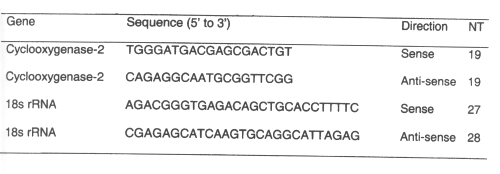
All of the chemotherapeutic agents tested increased gastroduodenal permeability as measured by sucrose excretion from baseline controls. The sucrose excretion in urine also appears to be dose-dependent for 5-fluorouracilbetween 200 and 400 mg/kg. The NSAID indomethacin at doses of 10-20 mg/kg significantly increased gastrodudenal permeability as measured by sucrose excretion in urine. (Figure 1)
Figure 1: Urinary excretion of sucrose after administration of single doses of indomethacin and chemotherapeutic agents to rats. (N=5, Mean+/- SEM).
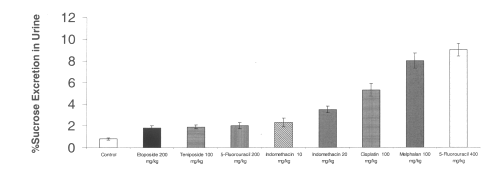
The chemotherapeutic agents tested all increased intestinal permeability from baseline controls. The excretion of 51Cr-EDTA in urine also appears to be dose-dependent for 5-fluorouracil between 200 and 400 mg/kg. Indomethacin at a dose of 10 or 20 mg/kg significantly increased intestinal permeability as measured by 51Cr-EDTA excretion in urine. (Figure 2)
Figure 2: Urinary excretion of 51Cr-EDTA after administration of single doses of indomethacin and chemotherapeutic agents to rats. (N=5, Mean+/- SEM).
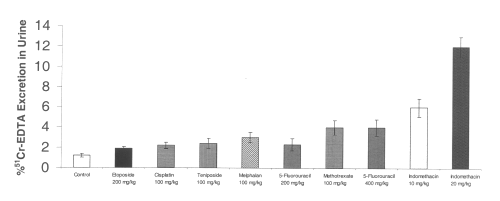
All of the chemotherapeutic agents tested also increased cecal hemoglobin from baseline controls. Indomethacin at a dose of 10 mg/kg significantly increased gastrointestinal bleeding as measured by cecal hemoglobin. (Figure 3)
Figure 3: Cecal hemoglobin level after administration of single doses of NSAIDs and chemotherapeutic agents to rats. (N=5, Mean+/- SEM).
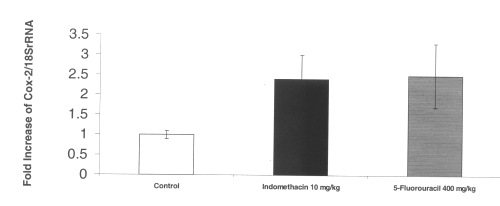
The RT-PCR of COX-2 mRNA in intestinal tissue demonstrated a 2-fold increase in up regulation of this inflammatory gene after administration of both indomethacin and 5-fluorouracil. (Figure 4)
Figure 4: COX-2 mRNA gene expression in rat intestinal tissue after administration of single doses of indomethacin and chemotherapeutic agents. (N=5, Mean+/- SEM).
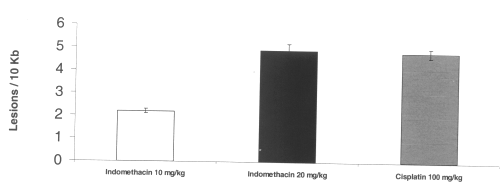
Mitochondrial DNA damage was apparent in gastric and intestinal tissue. Dose-dependent mitochondrial DNA damage was induced by indomethacin in the stomach. Cisplatin also induced mitochondrial oxidative DNA damage. (Figure 5)
Figure 5: Mitochondrial DNA damage in rat gastric tissue after administration of single doses of indomethacin and cisplatin. (N=5, Mean+/- SEM).
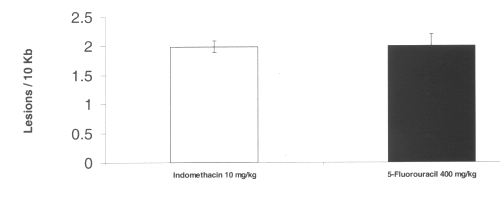
Treatment with both indomethacin and 5-fluorouracil also caused mitochondrial oxidative damage in the intestine. No nuclear DNA damage was detected by any agent tested. (Figure 6)
Figure 6: Mitochondrial DNA damage in rat intestinal tissue after administration of single doses of indomethacin and 5-fluorouracil. (N=5, Mean+/- SEM).
DISCUSSION
The rat appears to be a suitable model for examining gastrointestinal permeability to chemotherapeutic agents at toxic doses. Similar to our previous findings in the rat with NSAIDs the toxic response in the rat to chemotherapy from various classes is dose-dependent.(17-18) Sucrose excretion in urine effectively detected gastroduodenal permeability and is a surrogate marker of gastroduodenal damage whereas 51Cr-EDTA excretion in urine reflected small intestinal permeability and correlated with enteropathy. The present study utilized a commercially available assay kit for sucrose detection in urine as opposed to previous report (18) where indomethacin at 10 mg/kg did not reach statistical significance.
It has been previously demonstrated that NSAIDs inhibit mitochondrial oxidative phosphorylation and that this may precede changes in gastrointestinal permeability.(36-37) The current results (Figures 5 and 6) confirm that mitochondrial damage is an early event in the pathogenesis of gastric toxicity due to both NSAIDs and cisplatin. This damage is specific to the mitochondria rather than nuclear cellular DNA and parallels the results previously demonstrated with cisplatin in leukemia cells (38). Interestingly, significant mitochondrial damage occurs without nuclear DNA damage. This may be a consequence of the lack of a compact nucleosome structure as compared with the nuclear DNA, limited mitochondrial repair pathways, as well as the proximity of the mitochondrial DNA to the main source of reactive oxygen species generated. (34-35) More work is needed to understand the molecular events underlying the susceptibility of the mitochondrial DNA to oxidative damage. A sequela of these early events of gastrointestinal permeability is the up regulation of COX-2 in the intestine (39). Our results also confirm up-regulation of COX-2 mRNA is evident in indomethacin and 5-fluorouracil enteropathy. When macroscopic damage to the intestine is evident, damage to mitochondrial DNA also appeared to be present. The pathogenic time course and specific sequence of events leading to these toxic insults requires further study.
An increasing body of knowledge of chemotherapeutic related side-effects in the literature suggests the importance of determining gastrointestinal integrity following drug treatment and in various diseases.(9,20,40) Given the well documented changes in gastrointestinal permeability and the involvement of mitochondria and oxidative stress in the gastrointestinal tract, using the rat as a model to study gastrointestinal chemotherapeutic side-effects may help us in understanding the mechanism of action of anti-neoplastic drugs in producing GI abnormalities. The rat appears to be a suitable animal model to study these GI disturbances as it responds in a similar fashion to the changes described in humans. Further studies are ongoing in our laboratory to examine in detail the pathogenic sequences involved in the process of gastrointestinal damage between drug, dose and concentration required to elicit these effects.
ACKNOWLEDGMENTS
American Cancer Society Institutional Research Grant to NMD. A SURF fellowship from Merck-Frosst and ASPET to JAY. Dr. B Van Houten, Dr. T Schmittgen and Brian Zakrajsek for assistance with setting up the PCR techniques in Dr. Davies laboratory.
References
Blumberg D, Ramanathan RK. Treatment of colon and rectal cancer. J Clin Gastroenterol 2002; 34:15-26.
Ilson DH. New developments in the treatment of esophageal cancer. Curr Oncol Rep 2002; 4:213-221.
Shah MA. Recent developments in the treatment of gastric carcinoma. Curr Oncol Rep 2002; 4:193-201.
Magne N, Marcy PY, Chamorey E, Guardiola E, Pivot X, Schneider M, Demard F, Bensadoun RJ. Concomitant twice-a-day radiotherapy and chemotherapy in unresectable head and neck cancer patients: A long-term quality of life analysis. Head Neck 2001; 23:678-682.
Denham JW, Abbott RL. Concurrent cisplatin, infusional fluorouracil, and conventionally fractionated radiation therapy in head and neck cancer: dose-limiting mucosal toxicity. J Clin Oncol 1991; 9:458-463.
Bensadoun RJ, Magne N, Marcy PY, Demard F. Chemotherapy- and radiotherapy-induced mucositis in head and neck cancer patients: new trends in pathophysiology, prevention and treatment. Eur Arch Otorhinolaryngol 2001; 258:481-487.
Davies NM, Saleh JY, Skjodt NM. Detection and prevention of NSAID-induced enteropathy. J Pharm Pharm Sci 2000; 3:137-155.
Reuter BK, Davies NM, Wallace JL. Nonsteroidal anti-inflammatory drug enteropathy in rats: role of permeability, bacteria, and enterohepatic circulation. Gastroenterology 1997; 112:109-117.
Davies NM. Non-steroidal anti-inflammatory drug-induced GI permeability Aliment Pharmacol Ther 1998; 12:303-320.
Hanif R, Pittas A, Feng Y, Koutsos MI, Qiao L, Staiano-Coico L, Shiff SI, Rigas B. Effects of nonsteroidal anti-inflammatory drugs on proliferation and on induction of apoptosis in colon cancer cells by a prostaglandin-independent pathway. Biochem Pharmacol 1996; 52:237-245.
Mahmud T, Rafi SS, Scott DL, Wrigglesworth JM, Bjarnason I. Nonsteroidal antiinflammatory drugs and uncoupling of mitochondrial oxidative phosphorylation. Arthritis Rheum 1996; 39:1998-2003.
Melichar B, Kohout P, Bratova M, Solichova D, Kralickova P, Zadak Z. Intestinal permeability in patients with chemotherapy-induced stomatitis. J Cancer Res Clin Oncol 2001; 127:314-318.
Parrilli G, Iaffaioli RV, Capuano G, Budillon G, Bianco AR. Changes in intestinal permeability to lactulose induced by cytotoxic chemotherapy. Cancer Treat Rep 1982; 66:1435-1436.
Keefe DM, Cummins AG, Dale BM, Kotasek D, Robb TA, Sage RE. Effect of high-dose chemotherapy on intestinal permeability in humans. Clin Sci (Lond) 1997; 92:385-389.
Mansi J, Ellis E, Viner C, Mundy J, Smith T, Millar J, Milan S, Gore M, Cunningham D. Gut protection by cyclophosphamide "priming" in patients receiving high-dose melphalan--effect of drug scheduling. Cancer Chemother Pharmacol 1992; 30:149-151.
Fazeny-Dorner B, Veitl M, Wenzel C, Brodowicz T, Zielinski C, Muhm M, Vogelsang H, Marosi C. Alterations in intestinal permeability following the intensified polydrug-chemotherapy IFADIC (ifosfamide, Adriamycin, dacarbazine). Cancer Chemother Pharmacol 2002; 49:294-298.
Davies NM, Wright MR, Jamali F. Antiinflammatory drug-induced small intestinal permeability: the rat is a suitable model. Pharm Res 1994; 11(11): 1652-1656.
Davies NM, Corrigan BW, Jamali F. Sucrose urinary excretion in the rat measured using a simple assay: a model of gastroduodenal permeability. Pharm Res 1995; 12:1733-1736.
Meddings JB, Gibbons I. Discrimination of site-specific alterations in GI permeability in the rat. Gastroenterology 1998; 114:83-92.
Bjarnason I, MacPherson A, Hollander D. Intestinal permeability: an overview. Gastroenterology 1995; 108:1566-1581.
Ford J, Martin SW, Houston JB. Assessment of intestinal permeability changes induced by nonsteroidal anti-inflammatory drugs in the rat. J Pharm Toxicol Meth 1995; 34:9-16.
Chevreau N, Wang Y, Funk-Archuleta M. Effect of diets on 5-fluorouracil and cyclophosphamide toxicity.Nutr Cancer. 1995; 23: 205-220.
Tavakkolizadeh A, Shen R, Abraham P, Kormi N, Seifert P, Edelman ER, Jacobs DO, Zinner MJ, Ashley SW, Whang EE.Glucagon-like peptide 2: a new treatment for chemotherapy-induced enteritis. J Surg Res. 2000; 91: 77-82.
Robinson BA, Clutterbuck RD, Millar JL, McElwain TJ. Epidermal growth factor (hEGF) has no effect on murine intestine epithelial damage and regeneration after melphalan. Br J Cancer. 1985; 52: 733-737.
Fuskevag OM, Kristiansen C, Lindal S, Aarbakke J.Maximum tolerated doses of methotrexate and 7-hydroxy-methotrexate in a model of acute toxicity in rats. CancerChemotherPharmacol.2000; 46(1):69-73.
Fuskevag OM, Kristiansen C, Olsen R, Aarbakke J, Lindal S.Microvascular perturbations in rats receiving the maximum tolerated dose of methotrexate or its major metabolite7-hydroxymethotrexate.UltrastructPathol.2000;24:325-332.
Takahashi N, Kadota T, Kawano S, Ohta K, Ishikawa K, Kuroyanagi K, Hamajima Y, Ohta S, Kai S, Kohmura H. (Toxicity studies of VP 16-213 (II)--Oral one-month subacute toxicity in rats) J Toxicol Sci. 1986; 11 Suppl 1:17-49.
Takahashi N, Kadota T, Kawano S, Ishikawa K, Kuroyanagi K, Hamajima Y, Ohta K, Ohta S, Kai S, Kohmura H (Toxicity studies of VP 16-213 (I)--Acute toxicity in mice, rats and rabbits)J Toxicol Sci. 1986 Apr;11 Suppl 1:1-16.
Stahelin H.Delayed toxicity of epipodophyllotoxin derivatives (VM 26 and VP 16-213), due to a local effect. Eur J Cancer. 1976; 12:925-931.
Broomhead JA, Fairlie DP, Whitehouse MW. Cis-Platinum (II) amine complexes: some structure-activity relationships for immunosuppressive, nephrotoxic and gastrointestinal (side) effects in rats. Chem Biol Interact. 1980; 31(1):113-132.
Hartl A, Guttner J, Horn U, Jelinek F, Stockel U, Schroer HP, Hoffmann H. (Acute and subacute toxicity of antineoplastic Pt(II) and Pt(IV) coordination compounds in laboratoryrodents).ArchGeschwulstforsch.1989;59: 239-244.
Nakano K, Ike O, Wada H, Hitomi S, Amano Y, Ogita I, Nakai N, Takada K.Oral sustained-release cisplatin preparation for rats and mice. J Pharm Pharmacol.1997; 9: 485-490.
Tirmenstein MA, Nicholls-Grzemski FA, Schmittgen TD, Zakrajsek BA, Fariss MW. Characterization of nitric oxide production following isolation of rat hepatocytes. Toxicol Sci 2000; 53:56-62.
Torres SA, Chen Y, Svoboda T, Rosenblatt J, Van Houten B. Analysis of gene-specific DNA damage and repair using quantitative polymerase chain reaction. Methods 2000; 22:135-147.
Santos JH, Mandavilli BS, Van Houten B. Measuring oxidative mtDNA damage and repair using quantitative PCR. Methods Mol Biol 2002; 197:159-176.
Somasundaram S, Sigthorsson G, Simpson RJ, Watts J, Jacob M, Tavares IA, Rafi S, Roseth A, Foster R, Price AB, Wrigglesworth JM, Bjarnason I. Uncoupling of intestinal mitochondrial oxidative phosphorylation and inhibition of cyclooxygenase are required for the development of NSAID-enteropathy in the rat. Aliment Pharmacol Ther 2000; 14:639-650.
Somasundaram S, Rafi S, Hayllar J, Sigthorsson G, Jacob M, Price AB, Macpherson A, Mahmod T, Scott D, Wrigglesworth JM, Bjarnason I. Mitochondrial damage: a possible mechanism of the "topical" phase of NSAID induced injury to the rat intestine. Gut 1997; 41:344-353.
Kalinowski DP, Illenye S, Van Houten B. Analysis of DNA damage repair in murine leukemia L1210 cells using a quantitative polymerase chain reaction assay. Nucleic Acids Res 1992; 20(13): 3485-3494.
Tanaka A, Hase S, Miyazawa T, Takeuchi K. Up-regulation of cyclooxygenase-2 by inhibition of cyclooxygenase-1: a key to nonsteroidal anti-inflammatory drug-induced intestinal damage. J Pharmacol Exp Ther 2002; 300:754-761.
Smale S, Bjarnason I. Determining small bowel integrity following drug treatment. Determining small bowel integrity following drug treatment. Br J Clin Pharmacol. 2003; 56(3): 284-291.
Corresponding Author: Neal M Davies, College of Pharmacy, Department of Pharmaceutical Sciences, Washington State University, Pullman, Washington, 99164-6534, USA. ndavies@wsu.edu
Published by the Canadian Society for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences.
http://www.ualberta.ca/~csps