J Pharm Pharmaceut Sci (www.ualberta.ca/~csps) 6(3):352-359, 2003
A comparison of gastrointestinal permeability induced by diclofenac-phospholipid complex with diclofenac acid and its sodium salt.
Tahereh Khazaeinia1, Fakhreddin Jamali
Faculty of Pharmacy and Pharmaceutical Sciences, University of Alberta, Edmonton, Alberta, Canada T6G 2N8Received 17 September 2003, Revised 8 December 2003, Accepted 9 December 2003
PDF version
Abstract
PURPOSE: Gastrointestinal (GI) side effects of the nonsteroidal anti-inflammatory drug (NSAID) diclofenac may be reduced if it is administered as a complex with phospholipid. The upper and lower GI permeability induced by a diclofenac-dipalmitoyl phosphatidyl choline (DPPC) complex were compared with those of diclofenac acid and its sodium salt in rats. METHODS: Pharmacokinetic studies were carried out to assess bioavailability of diclofenac preparations. Adult male Sprague-Dawley rats were dosed orally (equivalent to 15 mg/kg diclofenac sodium) as the acid or its sodium salt as well as diclofenac-DPPC complex. Upper and lower GI permeability, as surrogate markers of toxicity were determined using sucrose and 51Cr-EDTA, respectively. RESULTS: At 1 h post-dose only diclofenac sodium induced a significant increased upper GI permeability. Three h post-dose all formulations significantly increased upper GI permeability although the diclofenac acid had the least effect. In the lower GI tract, the induced increase in permeability was significant at 1 and 3 h post-dose for all formulations with no significant differences between them. CONCLUSION: The induced upper and lower GI toxicity of diclofenac was formulation and time dependent. The lack of effect of diclofenac acid was due to the decreased availability of the drug. In the upper GI tract, up to 1 h post-dose, the diclofenac-DPPC complex demonstrated reduced upper gastroduodenal permeability as measured by sucrose. However, the protective effect of DPPC did not last and was not extended to the lower GI tract due to the systemic effect, contribution from the enterohepatic recirculation and/or dissociation of the complex. In assessing diclofenac GI toxicity, the effect of the different formulations on the entire GI tract at various times after drug administration must be considered.
Introduction
Since the introduction of cyclooxygenase-2 selective drugs, the use of conventional non-steroidal anti-inflammatory drugs (NSAIDs) has declined. Nevertheless, the latter are still very much in use. Diclofenac is one of the most widely prescribed NSAIDs for its analgesic, and anti-inflammatory indications. It is also available as an over the counter product in some countries. Similar to other NSAIDs, diclofenac use is associated with rare, but serious and sometimes fatal, gastrointestinal (GI) side effects, including ulceration, and hemorrhage, especially in the elderly (1, 2).
Different approaches have been taken to decrease NSAID-induced GI toxicity. For example, incorporation of NSAIDs with phospholipid has been suggested to improve GI safety of these drugs (9). The presence of an adsorbed layer of surface-active phospholipids on the surface of the mucus that covers the surface epithelium is suggested to protect the GI tissues by providing a hydrophobic layer between the epithelium and the luminal contents (10, 11). In addition, the phospholipid layer increases mucosal resistance to luminal acidity by repelling the diffusion of hydrogen ions (12). There are a number of lipids that appear to be covalently and non-covalently associated with mucus glycoprotein (9, 13, 14). Phosphatidylcholine with the dipalmitoyl species represents the most potent surface-active phospholipid (11). It has been suggested that ionic binding between DPPC and an NSAID shields the NSAID from pH-dependent changes hence the complex remains lipophilic even as the intragastric pH approaches neutrality (15).
Although, it has been reported that NSAIDs associated with zwitterionic phospholipids may reduce GI toxicity (9), there is not sufficient data on the pharmacokinetics of NSAIDs administered as these preparations. This is important since phospholipid may influence the absorption characteristics of the drug (19).
Previous studies have demonstrated that GI toxicity of diclofenac is due to both local as well as systemic effects and enterohepatic recirculation (20). Therefore, due to contributions of local effects, modification of diclofenac formulations may reduce GI toxicity. The objectives of this study were to compare GI toxicity of diclofenac administered as diclofenac acid, the sodium salt, and a phospholipid complex. This was achieved by studying the pharmacokinetics and the pattern of increased upper and lower GI permeability caused by the latter formulations. Permeability changes were used as the surrogate marker of NSAID induced GI toxicity in the rat model (7, 4). Sucrose (4) and 51Cr-ethylendiaminetetraacetic acid (51Cr-EDTA) (7) offer non-invasive approaches for assessment of NSAID-induced upper and lower GI tract, respectively (3-6, 7).
Materials And METHODS
Chemicals
Dipalmitoyl phosphatidyl choline (DPPC) was purchased from Amagasaki (Hyogo, Japan). Diclofenac sodium powder, naphthoxy acetic acid sodium, cetyl trimethylammonium bromide and Trinder's reagent were from Sigma (St. Louis, MO, USA). Methylcellulose, D-glucose, and potassium dihydrogen orthophosphate were acquired from BDH Chemicals (Edmonton, Canada) and sucrose was from Aldrich Chemical Company Inc (Milwaukee, WI, USA). 51Cr-EDTA was obtained from Dupont NEN (Wilmington, DE, USA). Methoxyflurane was purchased from Janssen Pharmaceutica (North York, Canada). All solvents and reagents were of HPLC and analytical grade, respectively.
Formulations
Diclofenac acid was prepared by acidification of an aqueous solution of diclofenac sodium, extraction into chloroform, and subsequent recrystallization. Diclofenac-DPPC complex was prepared by associating diclofenac acid with an equimolar concentration of DPPC. A suspension of the complex in water was used as the diclofenac-DPPC complex. Diclofenac sodium and diclofenac acid suspension in 1% methylcellulose were used as diclofenac sodium and diclofenac acid preparations, respectively. Diclofenac content of all formulations was confirmed by HPLC.
Diclofenac Assay
Plasma concentrations of diclofenac sodium were quantified using a reverse phase HPLC method at ambient temperature. Briefly, 100 ml of rat plasma was mixed with 25 ml of internal standard (naphthoxy acetic acid sodium, 10 mg/ml), 10ml of phosphoric acid and 1 ml chloroform. The contents of the tube were vortexed for 1 min, and then centrifuged at 1800 x g for 5 min. Following separation and evaporation of the organic layer, the residue was reconstituted in 200 ml of HPLC water and 50-100 ml of the aqueous phase was injected into the HPLC system.
The HPLC system consisted of a M-45 solvent delivery system (Waters Associate, Inc., Milford, MA, USA), a SPD-10A UV-Vis variable wavelength detector (Shimadzu Coroperation, Kyoto, Japan), a SIL-9A autoinjector (Shimadzu analytical instruments division, Kyoto, Japan), and a 10 cm x 4.6 mm I.D. partisil ODS-3 analytical column (Whatman Inc, Clifton, NJ, USA). The mobile phase consisted of acetonitrile-phosphate buffer (33:67 v/v). Phosphate buffer was prepared by dissolving 6.8 g KH2PO4 (0.05M), 1 ml triethanolamine, and 3 ml 2 M sulfuric acid in 1000 ml water (pH 4.5). The flow rate was 1ml/min and detection was performed at 276 nm.
Under the chromatographic conditions employed, the internal standard and diclofenac eluted at 5.5 and 21 min, respectively. A linear concentration-response relationship was found within 0.025-15 mg/ml (r2>0.999) range. The assay was suitable for analysis of plasma samples (0.1 ml) with an acceptable coefficient of variation (<10%) and sensitivity (25 ng/ml).
Differential Scanning Calorimetry (DSC)
Thermograms of diclofenac acid, diclofenac sodium and the diclofenac-DPPC complex were recorded using a Seiko SSC/5200 differential scanning calorimeter (Plymouth Meeting, Pennsylvania, USA). The thermal behavior was studied by heating 2.0±0.2 mg of each individual sample in a covered sample pan under nitrogen gas flow. The investigations were carried out over the temperature range 25-200° with a heating rate of 10°/min.
Animals
Male Sprague- Dawley rats (250-300 g) were housed at ambient temperature in individual metabolic cages (Fisher Scientific, Edmonton, Canada). The experiments were approved by the Animal Care Committee of the University of Alberta.
Pharmacokinetic Study
Male Sprague-Dawley rats (n=5/group) were anesthetized with methoxyflurane. A PE-10 tubing fitted into a PE-50 catheter was implanted into the right jugular vein for the collection of blood samples. Rats received, via a gastric gavage, single oral doses of diclofenac sodium, diclofenac acid suspension in 1% methylcellulose or diclofenac -DPPC complex suspension in water (equivalent to 15 mg/kg of diclofenac sodium). They were fasted overnight and during the experiment but had free access to water. Blood samples (0.2 ml) were taken at 0, 0.08, 0.17, 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, and 8 h post-dose, plasma was separated, and stored at -20° until analysis.
Areas under the plasma concentration-time curve (AUC) of 0-1 h, 0-3 h and 0-8 h post-dose periods were calculated using the linear trapezoidal rule. Peak plasma concentration (Cmax ) and its time of attainment (tmax ) were recorded without curve fitting.
GI permeability Study
Rats (n=6/group) were dosed orally with diclofenac sodium, diclofenac acid, or diclofenac acid-DPPC complex (equivalent to 10 mg/kg diclofenac sodium). Control rats in each group received placebo (either DPPC or 1% methylcellulose, as appropriate). At 1, and 3 h post-dose which respectively coincide with the maximum increased permeability for sucrose (4) and 51Cr-EDTA (7), 1 ml of the permeability marker containing 1 g sucrose and 10m Ci 51Cr EDTA was dosed by oral gavage.
Relative permeability was assessed by calculating the percent of the administered dose of the probes excreted in urine up to 8 h. Upper and lower GI permeability changes were expressed as percent increases as compared with control rats. Sucrose and 51Cr-EDTA were measured using a spectrophotometer (4) and gamma counter (7), respectively.
Statistical analysis
Differences between two means were determined using the unpaired Student's t-test. One way ANOVA followed by Duncan's New Multiple Range tests were used to assess differences between more than two means. A p value of <0.05 was considered significant. Data are presented as mean ± standard error.
Results
Differential Scanning Calorimetry
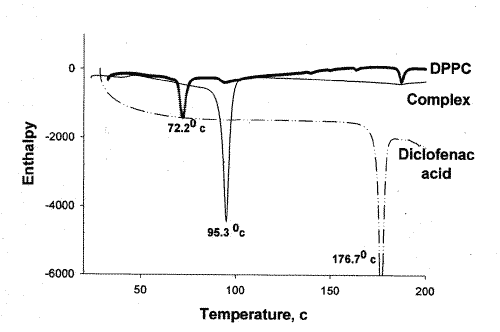
In order to substantiate the association of diclofenac acid with DPPC, DSC analysis was performed on diclofenac acid, DPPC, and the diclofenac-DPPC complex. The results of the DSC test confirmed the association of diclofenac acid and DPPC in the complex as both peaks representing diclofenac acid and DPPC changed position (Figure 1).
Figure 1: DSC thermograms of diclofenac acid, DPPC, and diclofenac acid-DPPC complex.
Diclofenac acid concentration per milligram of complex measured using HPLC indicated no degradation of the acid in the complex.
Pharmacokinetics
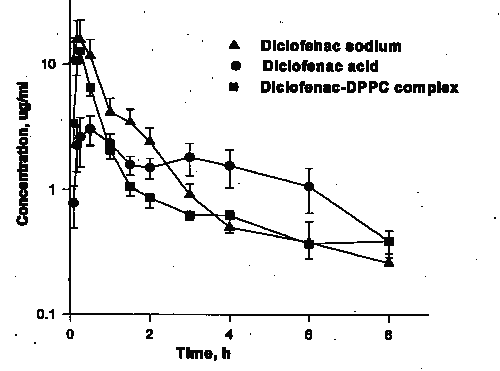
There were no significant differences in the AUC0-8 values for the three preparations (Table 1, Figure 2).
Table 1: Bioavalability indices of diclofenac acid, diclofenac sodium and diclofenac-DPPC complex following oral administration of 15 mg/kg (diclofenac sodium equivalent) to rats.

a, Significantly different from other preparations.
Figure 2: Concentration vs. time plot following administration of diclofenac sodium, diclofenac acid, and diclofenac -DPPC complex. (N=5/group, mean ± SE).
Diclofenac acid, however, was absorbed slower than other two formulations as indicated by its significantly longer Tmax, and lower Cmax as well as smaller AUC0-1. A comparison of AUC0-3 with that of AUC0-8 reveals that the absorption of the sodium salt, however, was 90% complete in 3 h so that its AUC0-3 was significantly greater than other preparations.
GI Permeability
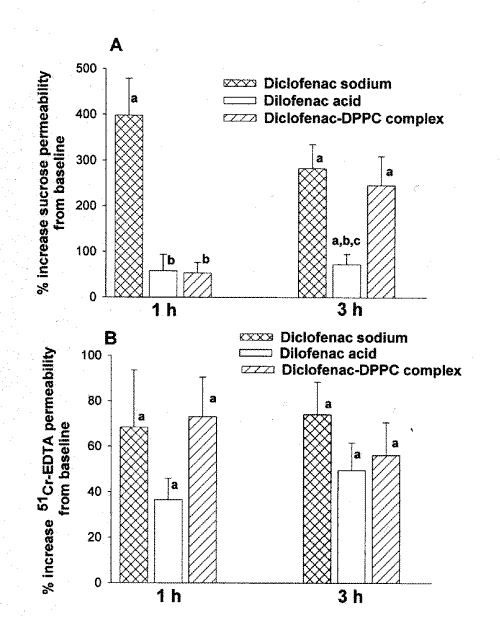
As depicted in Figure 3A, at 1 h post-dose, neither diclofenac acid nor diclofenac-DPPC complex significantly increased permeability of sucrose.
Figure 3: Percent increased upper (A) and lower (B) GI permeability at 1, and 3 h post-dose following administration of diclofenac sodium, diclofenac acid, and diclofenac-DPPC complex. (a, significantly different from baseline. b, significantly different from diclofenac sodium. c, significantly different from diclofenac-DPPC complex). (n=6/group, mean ± SE).
Diclofenac sodium, on the other hand, caused a significant elevation of the upper GI permeability. At 3 h post-dose, all formulations significantly increased upper GI permeability but the induced toxicity by diclofenac sodium and diclofenac-DPPC complex significantly exceeded that of diclofenac acid (Figure 3A).
In the lower GI tract, increased induced permeability was significant at both 1, and 3 h post-dose for all three formulations (Figure 3B), and no significant difference was observed between the examined formulations.
Discussion
NSAID therapy is commonly associated with GI tract side effects. Depending on the NSAID, the GI toxicity is due to either direct local and/or systemic effects. Direct local effects may be due to the local GI exposure after oral administration and also secondarily to biliary excretion into the GI tract. The post absorption systemic effect may be manifested following all routes of drug administration including parenteral (21) or rectal doses (22). Our previous studies demonstrated that diclofenac induced-GI toxicity is due to both local exposure and systemic distribution (20).
Many approaches have been used to inhibit or decrease the severity of the GI toxicity of NSAIDs. They include designing of prodrugs (23, 24), nitric oxide donor NSAIDs (28) and selective cyclooxygenase-2 inhibitors (29), as well as preparation of enteric coated and sustained release formulations (25, 26), cyclodextrin-NSAID (27), and dipalmitoyl-phosphatidylcholine-NSAID (9) complexes. Diclofenac is available on the market in enteric coated and sustained release formulations. Our previous data suggest that sustained-release formulations of diclofenac induces significantly increased intestinal permeability compared to immediate release preparations with no safety advantage in the upper GI tract (20). In addition, severe damage in the lower intestine has been reported following administration of sustained-release diclofenac formulations (30).
Diclofenac is a weak acid (pKa, 4.0) with a partition coefficient of 13 (31) in octanol/phosphate buffer (pH, 7.4). The solubility of diclofenac under physiological conditions ranges from 17.8 mg/L water at neutral pH to less than 1 mg/L at acidic pH (32, 33). Sodium diclofenac has a solubility of 1113 mg/L in water (33). The association of diclofenac with zwitterionic phospholipids, which may be both electrostatic and hydrophobic in nature, renders the phospholipids more water-soluble and the NSAID more lipid-soluble (34). It has been reported that the diffusion of NSAIDs across lipid membranes and into target cells is accelerated when it is present as a complex with DPPC (9). Our results revealed that both diclofenac sodium and diclofenac-DPPC complex exhibit higher initial plasma drug concentrations than diclofenac acid (Figure 2) as reflected in greater AUC values during the first one and three hours post-dose (Table 1).
It has been reported that NSAIDs have the ability to decrease the normal hydrophobic properties of the mammalian stomach (9). The gastric mucosa has non-wettable hydrophobic surface characteristics that protect the epithelium from the luminal acid (16). Hydrophobicity is found to be different in the esophagus, antrum, proximal and distal duodenum, and the colon (16). Furthermore, surface hydrophobicity in the stomach is higher than in the proximal duodenum (35). Hydrophobicity in the GI tract is, at least in part, due to appreciable quantities of phosphatidycholines and other surface-active phospholipids found in the mucosal surface of GI tract (16). Surface mucus cells have the capacity to synthesize, store and secrete phospholipids into the mucus gel layer, a process that can be modulated by prostaglandins (36). Phosphatidylcholines, including the palmitoyl derivative, represent a prominent component of mucus phospholipids, and its mucosal concentration appears to be associated with the integrity of the protective barrier (11, 9). NSAIDs appear to decrease mucosal hydrophobicity due, perhaps, to their ability to suppress prostaglandin synthesis. In addition, they may chemically associate with phospholipids and destabilize them from the mucus gel layer (9). Such a biophysical transition would increase the stomach's wettability and result in an increase in the back diffusion of luminal acid into the mucosa and the development of erosions. It has been reported that NSAIDs chemically pre-associated with zwitterionic phospholipids have limited interaction with intrinsic phospholipids (9, 15) hence can no longer make a complex with the intrinsic phospholipids of the mucus gel layer of the GI tract (9,15). This may have protective hydrophobic properties.
Among the preparations examined in this study diclofenac sodium showed the highest extent of increased upper GI tract permeability at 1 h post-dose (Figure 3A). The increased sucrose permeability was also evident at 3 h post-dose as well as for 51Cr-EDTA both at 1 and at 3 h after administration of diclofenac sodium. This overall increased permeability of both upper and lower GI tract coincides with the observed more rapid and complete absorption of the salt as compared with other preparations. Indeed, AUC0-1 and AUC0-3, which are indicative of the rate of absorption, are significantly greater for the sodium salt as compared with other formulations (Table 1). In addition, the small elevation of AUC between 3 and 8 h post-dose indicate that diclofenac sodium is mainly absorbed within 3 h. The more rapid absorption of the sodium salt is likely due to its high solubility hence its availability at the site of absorption. This, in turn, might have resulted in greater exposure of the GI tract to the drug and have given rise to increased permeability along the tract. Diclofenac acid, on the other hand, has a very low water solubility, which appears to limit its rate of absorption (Figure 2, Table 1). Lower exposure of the upper GI tract to dissolved diclofenac acid has likely resulted in a significantly smaller increase in permeability of sucrose as compared with diclofenac sodium (Figure 3A). In the more distal parts of the GI tract where the pH is rather alkaline, the acid is expected to convert to its salt with much greater solubility. Therefore, the tract is more exposed to the soluble drug hence there will be more absorption and also greater increased permeability. This may explain the initially low and then gradual appearance of the drug in plasma following administration of the acid (Figure 2, Table 1). The limited increased upper GI permeability (Figure 3A) coupled with comparable increased lower GI permeability (Figure 3B) following diclofenac acid as compared with the sodium salt also appears to be in line with the above explanation, i.e., less drug release in the upper and more in the lower GI tract following administration of the acid.
Similar to diclofenac acid, the DPPC complex exhibited limited increased sucrose permeability within the first h post-dose. This, however, cannot be attributed to less drug release from the complex, as the drug absorption from the complex is rapid and comparable with that of the sodium salt (Figure 2). The presence of the complex, therefore, might have provided a degree of mucosal protection. The protective effect, however, appears to be limited to the upper GI tract and only during the first h post-dose (Figure 3). The complex yielded increased permeability comparable to that of diclofenac sodium for sucrose 3 h post-dose and for 51Cr-EDTA at both examined times (Figure 3).
The limited mucosal protective effect of DPPC may be due to the presence of enzymes such as lipase in the GI tract that may dissociate the drug from the complex. Other explanations for limited mucosal protective effect of DPPC include systemic effect and/or enterohepatic circulation of diclofenac (37) after absorption thereby further exposing the GI tract to the drug. Subcutaneous administration of diclofenac to rats has been reported to significantly alter the surface hydrophobicity and reduce the amount of phospholipids in both stomach and duodenum 3 h post-dose (35). Furthermore, diclofenac does not appear to induce significant changes in gastric or duodenal surface hydrophobicity in bile duct ligated rats (35). It seems that with continuous enterohepatic recirculation of diclofenac, there are local interactions between the drug and surfactant phospholipids in the GI tract (35).
These results are consistent with previous studies suggesting formulation-dependent toxicity of NSAIDs in the rat model (20, 38-40). Our data also highlight the relative importance of considering formulation-dependent factors when evaluating the pharmacodynamic and toxicodynamic actions of NSAIDs (41, 42).
In conclusion, in the upper GI tract, up to 1 h post-dose, diclofenac-DPPC complex was devoid of significant increased permeability, a marker of mucosal toxicity. However, in the lower GI tract, the complex was not safer than diclofenac sodium. In assessing diclofenac-induced GI toxicity, the effect on the entire GI tract and at different post- administration times must be considered.
REFERENCES
Peloso PM., Strategies and practice for the use of nonsteroidal anti-inflammatory drugs. Scan. J. Rheumatol. 25 (suppl 105): 29-48, 1996.
Figueras A, Capella D, Castel JM, Laporte JR., Spontaneous reporting of adverse drug reactions to non-steroidal anti-inflammatory drugs. Eur. J. Clin. Pharmacol. 47: 297-303, 1994.
Meddings JB, Sutherland LR, Byles NI, Wallace JL., Sucrose: a novel permeability marker for gastroduodenal disease. Gastroenterology 104: 1619 –1626, 1993.
Davies NM, Corrigan BW, Jamali F., Sucrose urinary excretion in the rat measured using a simple assay: a model of gastroduodenal permeability. Pharm. Res. 12: 1733-1736, 1995.
Ford J, Martin W, Houston JB., Assessment of intestinal permeability changes induced by nonsteroidal anti-inflammatory drugs in the rat. J. Pharmacol. Toxicol. Met. 34: 9-16, 1995.
Riendeau D., Percival M.D., Gordon R., Greig G., Guay J., Mancini J., Ouellet M., Wong E., Xu L., Boyce S., Visco D., Girard Y., Prasit P., Zamboni R., Rodger I.W., Gresser M., Ford-Hutchingson A.W., Young R.N., Chan C.C., Etoricoxib (MK-0663): pre-clinical profile and comparison with other agents that selectively inhibit cyclooxygenase-2. J Pharmacol Exp Ther, 296(2):558-566, 2001.
Davies N.M., Wright M.R., Jamali F., Anti-inflammatory drug induced small intestinal permeability: The rat is a suitable model. Pharm. Res. 11: 1652-1656, 1994.
Meddings JB, Kirck D, Merle E, Olson DVM., Noninvasive detection of nonsteroidal anti-inflammatory drug-induced gastropathy in dogs. Am. J. Vet. Res. 56: 977-981, 1995.
Lichtenberger LM, Wang ZM, Romero JJ, Ulloa C, Perez JC, Giraud M-N, Barreto JC., Non steroidal anti-inflammatory drugs (NSAIDs) associate with Zwitterionic phospholipids: Insight into the mechanism and reversal of NSAID-induced gastrointestinal injury. Nature Med. 11: 154-158, 1995.
Goddard PJ, Kao YJ, Lichtenberger LM., Luminal surface hydrophobicity of canine gastric mucosa is dependent on a surface mucous gel. Gastroenterology 98: 361-370, 1990.
Lichtenberger LM, Graziani LA, Dial EJ, Butter BD, Hills BA., Role of surface-active phospholipids in gastric cytoprotection, Science 219: 1327-1329, 1983.
Hills BA, Kirwood CA., Gastric mucosal barrier to hydrogen ions imparted by gastric surfactant in vitro. Gut 33: 1039-1041, 1992.
Sloniany BL, Sarosiek J, Slomiany A., Gastric mucus and mucosal barrier. Dig. Dis Sci. 5:125-45, 1987.
Sloniany BL, Slomiany S., Role of mucus in gastric mucosal protection. J. physiol. Pharmacol. 42:141-161, 1991.
Lichtenberger LM, Ulloa C, Romero JJ, Vanous AL, Illich PA, Dial EJ., Nonsteroidal anti-inflammatory drug and phospholipid prodrugs: Combination therapy with antisecretory agents in rats. Gastroenterology 111: 990-995, 1996.
Hills BA, Butler BD, Lichtenberger LM., Gastric mucosal barrier: hydrophobic lining to the lumen of the stomach. Am. J. Physiol. 244: G561-8, 1983.
Lugea A, Mourrelle M, Guarner F, Domingo A, Salas A, Malagelada JR., Phosphatidylcholine as mediators of adaptive cytoprotection of the rat duodenum. Gastroentrology 107: 720-727, 1994.
Kivinen A, Vikholm I, Tarpila S. A film balance study of the monolayer-forming properties of dietary phospholipids and the interaction with NSAIDs on the monolayers. Nat. Med. 1:154-158, 1994.
Habib MJ, Phan MT, Owusu-Ababio G., Dissolution profiles of flurbiprofen in phospholipid solid dispersions. Drug Dev. Ind. Pharm. 24: 1077-1082, 1998.
Khazaeinia T, Jamali F., Formulation dependent gasterointestinal toxicity of diclofenac following administration of immediate and sustained release formulation in rats. Inflammopharmacology, in press, 2003.
Maliekal J, Elboim CM., Gastrointestinal complications associated with intramuscular ketorolac tromethamine therapy in the elderly. Ann. Pharmacother. 29: 698-701, 1995.
Ligumsky M, Sestieri M, Karmeli F, Zimmerman J, Okon E, Rachmilewitz D., Rectal administration of nonsteroidal anti-inflammatory drugs. Gastroenterology 98: 1245-1249, 1990.
Shanbhag V R, Crider AM, Gokhale R, Harpalani, Dick RM., Ester and amide prodrugs of ibuprofen and naproxen: synthesis, anti-inflammatory activity, and gastrointestinal toxicity. J. Pharmaceut. Sci. 81: 149-154, 1992.
Dandona P, Jeremy JY., Nonsteroidal anti-inflammatory drug therapy and gastric side effects. Does nabumetone provide a solution. Drugs 40 (suppl): 16-24, 1990.
Hoftiezer JW, Silvoso GR, Burks M, ivey KJ., Comparison of the effects of regular and enteric-coated aspirin on gastroduodenal mucosa of man. Lancet 2: 609-612, 1980.
Trondstad RI, Aadlland E, Holler T, Olaussen B., Gastroscopic findings after treatment with enteric-coated and plain naproxen tablets in healthy subjects. Scan. J. Gastroenterol. 20: 239-242, 1985.
Acerbi D, Bonati C, Boscarino G, Bufalino L, Cesari F, D’ Ambrosio E, Mansanti P, Scali G., Pharmacokinetic study on piroxicam at the steady-state in elderly subjects and younger adults after administration of piroxicam beta-cyclodextrin. Int. J. Clin. Pharmacol. Res. 8: 175-180, 1988.
Wallace JL, Reuter B, Cicala C, McKnight W, Grisham M, Cirino G., Novel nonsteroidal anti-inflammatory drug derivatives with markedly reduced ulcerogenic properties in the rat. Gastroenterology 107: 173-174, 1994.
Mitchell JA, Akarasereenont P, Thiemermann C, Flower RJ, Vane JR., Selectivity of nonsteroidal anti-inflammatory drugs as inhibitors of constitutive and inducible cyclooxygenase. Proc. Natl. Acad. Sci. 90: 11693-11697, 1993.
Choi VMF, Coates J, Thomson ABR, Russel AS., Small bowel permeability- a variable effect of NSAIDs. Clin. Invest. Med. 18:357-361, 1995.
Menasse R, Hedwall PR, Kraetz J, Pericin C, Riesterer L, Sallmann A, Ziel R, Jaques R., Pharmacological properties of diclofenac sodium and its metabolites. Scand. J. Rheumatol. 22: 5-16, 1978.
Chiarini A, Tartarini A, Fini A., pH-Solubility relationships and partition coefficients for some anti-inflammatory arylaliphatic acids. Arch. Pharm. 317: 268-273, 1984.
Fini A, Laus M, Orienti I, Zecchi V. Dissolution and partition thermodynamic functions of some anti-inflammatory drugs. J. Pharm. Sci. 75: 23-25, 1986.
Lichtenberger LM, Ulloa C, Romero JJ, Vanous AL, Romero JJ, Dial EJ, Illich PA, Walters ET., Zwitterionic phospholipids enhance aspirin’s therapeutic activity, as demonstrated in rodent model systems. J. Pharmacol. Exp. Ther. 277: 1221-1227, 1996.
Lugea A, Antolin M, Mourelle M, Guarner F, Malgelada JR., Deranged hydrophobic barrier of the rat gastroduodenal mucosa after parenteral nonsteroidal anti-inflammatory drugs. Gastroenterology. 112: 1931-1939, 1997.
Kao YC, Lichtenberger LM., Effect of 16, 16 dimethyl prostaglandin E2 on the lipid organells of rat gastric surface mucous cells. Gastroentrology. 104: 103-113, 1993.
Fukuyama T, Yamaoka K, Ohata Y, Nackagawa T., A new analysis method for disposition kinetics of enterohepatic circulation of diclofenac in rats. Drug. Metab. Dispos. 22: 479-485, 1994.
Khazaeinia T.; Jamali F., Evaluation of gastrointestinal of ibuprofen using surrogate markers in rats, effect of formulation, Clin. Exp. Rheumatol. 18:187—192, 2000.
Vakily M., Khorasheh F., Jamali F., Dependency of gastrointestinal toxicity on release rate of tiaprofenic acid: a novel pharmacokinetic-pharmacodynamic model. Pharm Res 16:123-129, 1999.
Davies N.M., Jamali F., Influence of dosage form on the gastroenteropathy of flurbiprofen in the rat: evidence of shift in the toxicity site. Pharm Res 14: 1597-1600, 1997.
Jamali F., Kunz-Dober C.M., Pain-mediated altered absorption and metabolism of dental surgery. Br J Clin Pharmacol 47: 391-396, 1999.
Lotsch J., Kettenmann B, Renner B, Drover D., Brune K., Geisslinger G., Kobal G., Population pharmacokinetics of fast release oral diclofenac in healthy volunteers: relation to pharmacodynamic in an experimental pain model. Pharm Res 17: 77-84, 2000.
Corresponding Author: T.Khazaeinia, tahereh_k@hotmail.com or F. Jamali, Faculty of Pharmacy and Pharmaceutical Sciences, University of Alberta, Edmonton, Alberta, Canada, T6G 2N8
Published by the Canadian Society for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences.
http://www.ualberta.ca/~csps