J Pharm Pharmaceut Sci (www.cspscanada.org) 7(4):22-28, 2004
Gelatin nanoparticles as a new and simple gene delivery system.
Klaus Zwiorek1, Julia Kloeckner2, Ernst Wagner2, Conrad Coester1*
1Department of Pharmacy, Pharmaceutical Technology and Biopharmaceutics; 2Department of Pharmacy, Pharmaceutical Biology-Biotechnology; Ludwig-Maximilians-University, Butenandtstr. 5-13, Munich, GermanyReceived 15 October 2004, Revised 3 January 2005, Accepted 3 January 2005, 03 February 2005
PDF Version
Abstract
PURPOSE: The aim of this study was to evaluate cationized gelatin nanoparticles as biodegradable and low cell toxic alternative carrier to existing DNA delivery systems. METHODS. Native gelatin nanoparticles were produced using a two step desolvation method. In order to bind DNA by electrostatic interactions onto the surface of the particles, the quaternary amine cholamine was covalently coupled to the particles. The modified nanoparticles were loaded with different amounts of plasmid in varying buffers and compared to polyethyleneimine-DNA complexes (PEI polyplexes) as gold standard. Transfection ability of the loaded nanoparticles was tested on B16 F10 cells. Additionally, the cell toxicity of the formulations was monitored. RESULTS. Different setups resulted in efficient gene delivery displayed by exponential increase of gene expression. The gene expression itself occurred with a certain delay after transfection. In contrast to PEI polyplexes, cationized gelatin nanoparticles almost did not show any significant cytotoxic effects. CONCLUSIONS. Cationized gelatin nanoparticles have shown the potential of being a new effective carrier for nonviral gene delivery. The major benefit of gelatin nanoparticles is not only the very low cell toxicity, but also their simple production combined with low costs and multiple modification opportunities offered by the matrix molecule.
Introduction
Safe and efficient delivery of DNA into cells is still a dominant task in today's biotechnological research. Direct injection of naked DNA is the easiest way of administration, but this approach is limited to only a few applications as for example injection into solid tumors (1) or muscle tissue (2). For most applications direct injection is not efficient enough due to enzymatic degradation and electrostatic repulsion between the negatively charged cell membrane and DNA, consequently inhibiting efficient cellular uptake.
For these applications a carrier system, either viral or nonviral, is needed. Statistics covering all clinical trials worldwide that are currently dealing with gene delivery reveal that viral vectors are still the clear number one, being used in more than 70% of all protocols (3). Nevertheless, having the advantage of the highest efficiency in delivery, viral vectors also have major drawbacks with respect to safety due to possibly severe immune reactions or oncogenic effects. These substantial reasons made the development of nonviral alternatives highly desirable. Common strategies are either complexes of nucleic acids with cationic polymers, i.e. polyplexes (4, 5), or based on cationic lipids, i.e. lipoplexes (4, 6). Furthermore, plasmid DNA can be loaded on the surface of previously formed solid nanoparticles as for example solid-lipid nanoparticles (7) or silica nanoparticles (8). But although a variety of efforts have been made in the past, the ideal system has not been found yet, since most of the physiologically well tolerated carriers lack in transfection efficiency and the majority of the efficient carriers are poor in terms of biodegradability or cell toxicity.
Our research group focuses on gelatin as potential basis for colloidal carrier systems. The water-soluble macromolecule is obtained by heat dissolution at alkaline or acidic conditions and partial hydrolysis of collagen in animal skins, bones and tendons. The majority of the tonnages produced every year are known to be utilized in food industries. But also the amount of gelatin applied in pharmaceutical industries is not negligible, especially as far as capsules and ointments are concerned (9). In terms of nanopharmaceutics, gelatin was already considered as interesting biodegradable base material in the early days of particle development (10). The interest was based on the facts that gelatin has been known for its low immunogenicity for many years (11) and is administered intraveneously since it is an ingredient of various registered blood substitutes. Despite the fact that a preparation method for gelatin nanoparticles using desolvation was already described in 1978 (10), the quality of nanoparticles being prepared by this procedure is rather unsatisfying. Resulting particles commonly have broad size distributions and tend towards instabilities. Finally, the reproducibility is inadequate as well. The underlying reason is the heterogeneity of gelatin having a broad molecular weight range (12, 13). A solution for these problems was the development of the two step desolvation technique (14) that allows the production of homogeneous and stable colloidal spheres. Since we established a surface modification of the nanoparticles leading to positively charged particles (15) and validated the stable nucleotide loading (16), the aim of this study was to test our formulations in vitro . Positive results of other groups that have already shown the potential of gelatin (17-19) and cationized gelatin (20-22) as complexing agent or hydrogel matrix for gene delivery encouraged our plans.
Besides the optimization of experimental conditions, a second focus of the study was to monitor the cell toxic effects of the system, to prove the thesis that gelatin nanoparticles are physiologically very well tolerated. PEI polyplexes, known for their high transfection efficiency (23, 24) were chosen as gold standard to rate the potential of the new nonviral carrier.
Materials and Methods
Materials
Gelatin type A from porcine skin (175 Bloom), glutaraldehyde (25%), 1-ethyl-3-(3-dimethyl-aminopropyl) carbodiimide hydrochloride (EDC) and (2-aminoethyl)-trimethylammoniumchloride hydrochloride (cholaminechloride hydrochloride) for the preparation of gelatin nanoparticles were purchased from Sigma-Aldrich (Taufkirchen, Germany). The acetone (p.a.) needed for this procedure was purchased from VWR International (Ismaning, Germany).
Linear PEI with an average molecular weight of 22 kDa (PEI 22) was purchased from Euromedex (Exgen 500, Euromedex, Souffelweyerheim, France).
The DNA plasmid used in our studies was pCMVLuc (Photinus pyralis luciferase under the control of the CMV enhancer/promoter) as described by Plank et al. (25) purified with the EndoFree Plasmid Kit from Qiagen (Hilden, Germany).
Preparation of cationized gelatin nanoparticles
Plain gelatin nanoparticles were produced by the two step desolvation technique (14). The first step is performed to discard low molecular weight components of gelatin which would make the production of stable nanoparticles with a uni-modal size distribution impossible. After resolvation of the sediment and pH-adjustment (pH 3.0), the in-situ particles were prepared by a second desolvation step. In order to bind DNA by electrostatic interactions onto the particles' surface, the quaternary amine cholamine was covalently coupled onto the surface. This step was a further optimization of procedures previously described by Coester (15). An aqueous dispersion of native nanoparticles was adjusted to pH 4.5 and a molar excess of cholamine was added under constant stirring. After a certain time EDC was added to activate free carboxyl groups on the particles to react with cholamine (as well). The final particles were characterized by sizing via dynamic light scattering and measurement of the zeta potential (z potential). The determinations of both parameters were conducted with a Zetamaster (Malvern Instruments, Worcestershire, UK). Each assigned size and corresponding polydispersity index was the mean of 10 subruns. The z potential was determined by the same instrument and calculated as the mean of 10 individual measurements.
Cell culture
Cell culture media, fetal calf serum (FCS) and antibiotics were purchased from Invitrogen GmbH (Karlsruhe, Germany). B16F10 cells were grown in Dulbeco's Modified Eagle's Medium (DMEM) (1.0 g glucose/L) supplemented with 10% FCS at 37°C in 5% CO2 humidified atmosphere.
Plasmid DNA loading of gelatin nanoparticles
Two different batches of cationized nanoparticles were loaded with different amounts of plasmid (0.33 - 1% [m/m]) in varying conjugation media to find optimized conditions.
The used conjugation media were phosphate buffered saline (PBS) 7.4 and PBS 7.0/water (1:1 ratio). In order to bind the DNA onto the nanoparticles, a dispersion of nanoparticles was pipetted to a solution of plasmid DNA (40 mg/mL) in buffer and mixed vigorously. After an incubation period of 2 min, 50 mL nanoparticle dispersion containing 200 ng DNA (luciferase plasmid) loaded in various plasmid to nanoparticle ratios was directly added to each well. The nanoparticles were centrifuged and complete DNA loading was verified by analyzing the supernatant for unbound DNA, UV-metrically at 260 nm wavelength (15).
Preparation of PEI polyplexes
For complex preparation, PEI was used at a 1 mg/mL working solution, neutralized with HCl.
DNA complexes were prepared by first diluting PEI 22 in HBS (Hepes buffered saline: 20 mM Hepes, 150 mM NaCl, pH 7.1). This PEI-conjugate buffer solution was pipetted to plasmid DNA diluted in HBS and mixed vigorously. Complexes were formed at a molar ratio of PEI nitrogen atoms to DNA phosphate (N/P) of 6.
Complexes were allowed to stand for at least 20 min at room temperature before use. The final concentration of DNA (luciferase plasmid) in complexes was 20 μg/mL.
Complex containing 200 ng pCMVLuc was added directly to each well.
Transfection
5.0 x 10 3 cells per well were seeded into 96-well plates (Gibco). After 16 h of incubation with the different formulations, the transfection media was removed and replaced by fresh media containing 10% FCS. After 24 h media was again replaced.
During transfection and for the following 24h 100 U/mL penicillin and 100 mg/mL streptomycin were added.
Luciferase assay
For luciferase detection, cells were washed once with PBS, lysed in 50 mL of Promega cell lysis solution (Promega, Mannheim, Germany), and assayed as previously described (26). Luciferase activity was determined from 20 μL samples of the lysate using the Luciferase Assay system (Promega); measurements were performed in a luminometer (Lumat LB9507, Berthold, Bad Wildbad, Germany).
Values are given as light units and represent the total luciferase activity per 5.0 x 10 3 cells as mean ± standard deviations of at least triplicates. 2 ng of recombinant luciferase (Promega) corresponded to 10 7 light units.
Cell viability
Compared to PEI polyplexes, cell viability studies of loaded and unloaded nanoparticles, were performed using a CellTiter-GloÔ Luminescent Cell Viability Assay (Promega, Mannheim, Germany) according to the manufacturer's instructions. This assay is based on quantification of the ATP content of viable cells.
Results and Discussion
To control the success of the surface modification using cholamine, we determined the surface charge of unmodified gelatin nanoparticles compared to cationized particles by measuring their z potentials at different pH values (Figure 1).
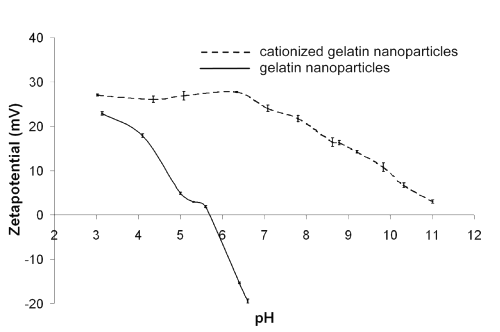
Figure 1: z potential measured in correlation to the pH value (ionic backround: 1 mM NaCl).
Whereas plain gelatin nanoparticles resulted in an isoelectric point of 5.7, the nanoparticles modified with cholamine remained positively charged within the measured pH range. Consequently, the pH of the physiological environment should not have any effect on the DNA loading.
Performing preliminary studies to determine the maximum amount of plasmid loading (data not shown), we found that we were able to load up to 50 mg plasmid per milligram nanoparticles onto the particles (5% [m/m]), but these formulations were very instable and agglomerated quickly in any physiological media. This rapid process could only be stopped by reducing the amount of loaded plasmid to 20 mg/mg (2% [m/m]). Nevertheless, within this study only the formulations loaded between 3.3 mg/mg and 10 mg/mg (0.33-1% [m/m]) showed significant transfection results. Searching for reasons, we investigated the z potentials of formulations containing different DNA to nanoparticle ratios (Figure 2).
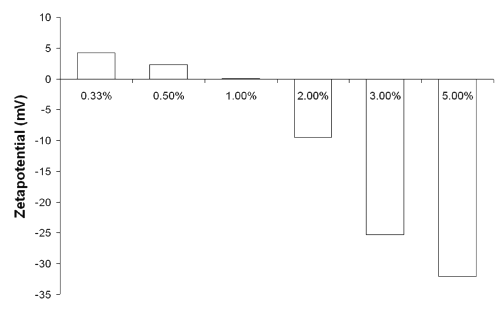
Figure 2: z potential determination of formulations with various ratios of DNA to nanoparticles (m/m).
The measurements were performed in physiological PBS 7.4. Formulations containing 3-5% (m/m) payload tended towards quick agglomeration and resulted in strong negative z potentials. Reducing the DNA content to 2% (m/m) prevented aggregations and correlated with a characteristic reduction of the formulations' z potential to -9.5 mV. Finally, the formulations that were able to transfect the B16 cells (0.33-1% [m/m]) had all either a neutral or slightly positive z potential, which appears to be crucial for successful gene delivery as it facilitates the passage through the negatively charged cell membrane. The two nanoparticle batches used in this study had a mean size of 288.3 nm (ZWgen288+) and 182.7 nm (ZWgen182+). Dynamic light scattering suggested a uni-modal size distribution for both batches, having low polydispersity indices of 0.067 (ZWgen288+) and 0.101 (ZWgen182+).
For nanoparticulate plasmid formulations loaded in PBS 7.4, none of the three different loading concentrations showed any significant gene expression after 24 h. However the PEI polyplexes showed their strongest gene expression (1.46 x 107 RLU [relative light units]) at this point (Figure 3).
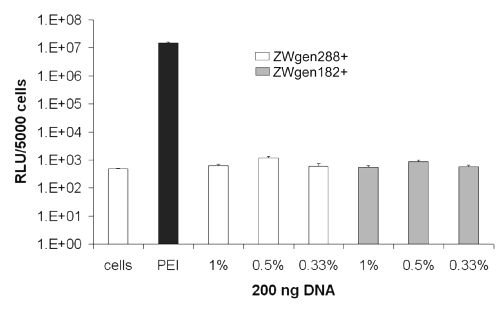
Figure 3: Transfection efficiency of cationized gelatin nanoparticles after 24 h; conjugation buffer: PBS 7.4; cells: untreated cells; PEI: standard polyplex; 1%, 0.5%, 0.33%: relative amount of plasmid loaded onto gelatin nanoparticles (m/m).
Performing the DNA loading of the particles in PBS 7.0/water (1:1 ratio) instead of PBS 7.4 led to first detectable gene expressions for the formulations loaded with 0.5% plasmid (m/m) after 24 h (Figure 4).
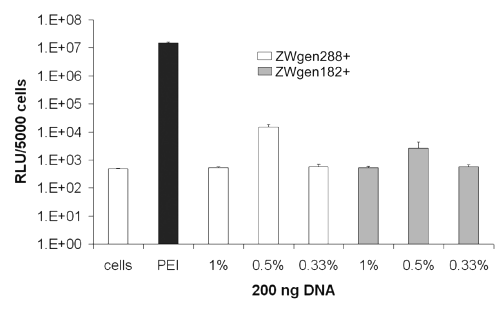
Figure 4: Transfection efficiency of cationized gelatin nanoparticles after 24 h; conjugation buffer: PBS 7.0/water (1:1 ratio); cells: untreated cells; PEI: standard polyplex; 1%, 0.5%, 0.33%: relative amount of plasmid loaded onto gelatin nanoparticles (m/m).
After 72 h, both the nanoparticles incubated in PBS 7.4 (Figure 5) and in PBS 7.0/water (1:1 ratio) (Figure 6) resulted in distinct increases of gene expression.
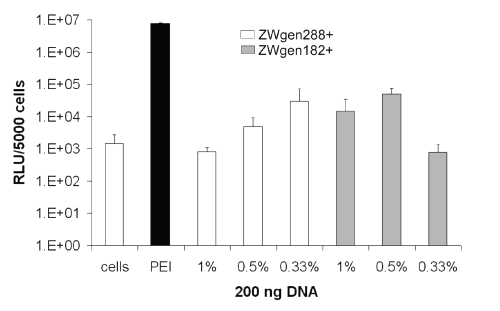
Figure 5: Transfection efficiency of surface-modified gelatin nanoparticles after 72 h; conjugation buffer: PBS 7.4; cells: untreated cells; PEI: standard polyplex; 1%, 0.5%, 0.33%: relative amount of plasmid loaded onto gelatin nanoparticles (m/m).
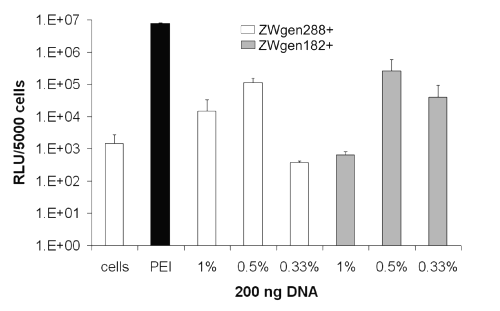
Figure 6: Transfection efficiency of surface-modified gelatin nanoparticles after 72 h; conjugation buffer: PBS 7.0/water (1:1 ratio); cells: untreated cells; PEI: standard polyplex; 1%, 0.5%, 0.33%: relative amount of plasmid loaded onto gelatin nanoparticles (m/m).
Formulations prepared in PBS 7.4 showed already noticeable outcomes with 2.98 x 104 RLU for the larger nanoparticles (ZWgen288+) containing 0.33% plasmid loading (m/m) and 4.94 x 104 RLU for ZWgen182+ (0.5% [m/m] plasmid). The best gene expressions could be achieved in PBS 7.0/water (1:1 ratio) with measured 1.12 x 105 RLU for ZWgen288+ and 2.63 x 105 RLU for ZWgen182 (both 0.5% [m/m] plasmid).
Overall, there is a tendency favoring the use of 182.7 nm large particles as carrier (ZWgen182+) with a relative amount of 0.5% plasmid (m/m) loaded onto the particles. As for the conjugation buffer, further studies have to be performed to find the ideal media. Compared to PEI, the peak gene expression occurs with a certain delay. One reason could be a prolonged intracellular processing due to slow endosomal escape or a different cellular uptake mechanism.
Since cytotoxicity of our formulations was the second focus of our experiments, we monitored the viability of the transfected B16 F10 cells simultaneously (Figure 7).
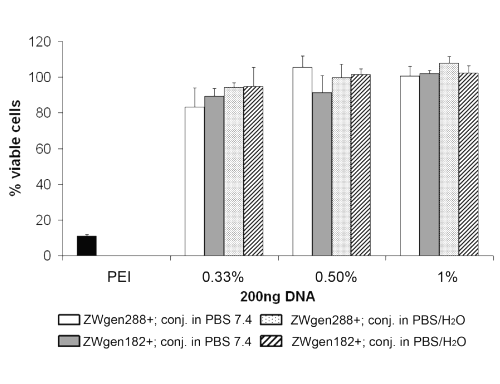
Figure 7: Cell viability of B16 F10 cells after 72 h of incubation; PEI: standard polyplex; 1%, 0.5%, 0.33%: relative amount of plasmid loaded onto gelatin nanoparticles (m/m).
After a total time of 72 h, only averaging 10.9% of the B16 F10 cells transfected with PEI polyplexes remained viable, whereas the viability of all cells that have been transfected with gelatin nanoparticle formulations was between 83.3 and 100%. For transfection, PEI formulations are typically incubated with cells for 4 h instead of 16 h as in this study. This can maybe explain the low cell viability of cells transfected with PEI polyplexes but also demonstrates the good physiological tolerance of gelatin nanoparticle formulations. A trend was observed, that formulations loaded with less relative plasmid amount are more toxic. The reason for this might be that the higher remaining positive net charge of the formulations induces lower cell viability. Furthermore, a marginal tendency towards formulations conjugated in PBS 7.4 showing less cell viability can be seen but in general, the cell toxicological effects of our formulations are minimal compared to PEI polyplexes. These results were also confirmed earlier in toxicological studies of our group that demonstrated the same very low in vitro toxicity compared to DOTAP/DOPC (1:1 ratio) liposomes (16).
Conclusion
A major benefit of gelatin nanoparticles for use in gene transfer is not only the very low cytotoxocity, but also their simple and reproducible production, which would facilitate future upscaling. Low costs of the base material gelatin make this approach commercially attractive. Gene transfer efficiency of our formulations are only approximately one order of magnitude lower than the efficiency of `gold standard' cationic PEI polyplexes; these in vitro transfection rates are significant and quite remarkable considering that we are still at an early stage of formulation optimization. One advantage offered by the amino acid side-chains of the gelatin matrix molecule is the option of multiple further modifications. This could be used for coupling of ligands to improve targeting diseased tissues, to enhance specific cellular uptake or affect intracellular distribution.
References
Vile, R. G., Hart, I. R., In vitro and in vivo targeting of gene expression to melanoma cells. Cancer Res, 53(5):962-967, 1993.
Wolff, J. A., Malone, R. W., Williams, P., Chong, W., Acsadi, G., Jani, A., Felgner, P. L., Direct gene transfer into mouse muscle in vivo. Science, 247(4949 Pt 1):1465-1468, 1990.
Gene therapy clinical trials worldwide, provided by J Gene Med , http://217.215.32.12/trials/index.html (updated July 1, 2004).
Felgner, P. L, Barenholz, Y., Behr, J. P., Cheng, S. H., Cullis, P., Huang, L., Jessee, J. A., Seymour, L., Szoka, F., Thierry, A. R., Wagner, E., Wu, G., Nomenclature for synthetic gene delivery systems. Hum Gene Ther, 8(5):511-512, 1997.
De Smedt, S.C., Demeester, J., Hennink, W.E., Cationic polymer based gene delivery systems. Pharm Res, 17:113-126, 2000.
Felgner, P. L., Nonviral strategies for gene therapy. Sci Am, 276(6):102-106, 1997.
Olbrich, C., Bakowsky, U., Lehr, C. M., Müller, R. H., Kneuer, C., Cationic solid-lipid nanoparticles can efficiently bind and transfect plasmid DNA. J Control Release, 77(3):345-355, 2001.
Kneuer, C., Sameti, M., Haltner E. G., Schiestl T., Schirra, H., Schmidt, H., Lehr, C. M., Silica nanoparticles modified with aminosilanes as carriers for plasmid DNA. Int J Pharm, 196:257-261, 2000.
Djagny, V. B., Wang, Z., Xu S., Gelatin: a valuable protein for food and pharmaceutical industries: review. Crit Rev Food Sci Nutr, 41(6):481-492, 2001.
[Marty, J. J., Oppenheim,R. C., Speiser, P., Nanoparticles - a new colloidal drug delivery system, Pharm Acta Helv, 53(1):17-23, 1978.
Schwick, H. G., and Heide, K., Immunochemistry and immunology of collagen and gelatin. Bibl Haematol, 33:111-125, 1969.
Farrugia, C. A., Groves, M. J., Gelatin behaviour in dilute aqueous solution: designing a nanoparticulate formulation. J Pharm Pharmacol, 51(6):643-649, 1999.
Fraunhofer, W., Winter, G., Coester, C., Asymmetrical flow field-flow fractionation and multiangle light scattering for analysis of gelatin nanoparticle drug carrier systems. Anal Chem, 76(7):1909-1920, 2004.
Coester, C. J., Langer, K., von Briesen, H., Kreuter, J., Gelatin nanoparticles by two step desolvation – a new preparation method, surface modifications and cell uptake. J Microencapsul, 17(2): 187-193, 2000.
Coester, C., Development of a new carrier system for oligonucleotides and plasmids based on gelatin nanoparticles. New Drugs, 1:14-17, 2003.
Zwiorek, K., Coester, C., Evaluation of surface-modified gelatin nanoparticles as delivery system for oligonucleotides and plasmids. Poster, 2003 AAPS Annual Meeting and Exposition, October 26 – 30, Salt Lake City, Utah, USA
Leong, K. W., Mao, H. Q., Truong-Le, V. L., Roy, K., Walsh, S. M., August, J. T., DNA-polycation nanospheres as non-viral gene delivery vehicles. J Control Release, 53(1-3):183-193, 1998.
[Truong-Le, V. L., August, J. T., Leong, K. W., Controlled gene delivery by DNA-gelatin nanospheres. Hum Gene Ther, 9(12):1709-1717, 1998.
Truong-Le, V. L., Walsh, S. M., Schweibert, E., Mao, H. Q., Guggino, W. B., August, J. T., Leong, K. W., Gene transfer by DNA-gelatin nanospheres. Arch Biochem Biophys, 361(1):47-56, 1999.
Hosseinkhani H., Aoyama T., Ogawa O., Tabata Y., Ultrasound enhancement of in vitro transfection of plasmid DNA by a cationized gelatin. J Drug Target, 10(3):193-204, 2002.
Fukunaka, Y., Iwanaga, K., Morimoto, K., Kakemi, M., Tabata, Y., Controlled release of plasmid DNA from cationized gelatin hydrogels based on hydrogel degradation. J Control Release, 80(1-3):333-343, 2002.
Kushibiki, T., Tomoshige, R., Fukunaka, Y., Kakemi, M., Tabata, Y., In vivo release and gene expression of plasmid DNA by hydrogels of gelatin with different cat-ionization extents. J Control Release, 90(2):207-216, 2003.
Boussif, O., Lezoualc'h, F., Zanta, M. A., Mergny, M. D., Scherman, D., Demeneix, B., Behr, J. P., A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc Natl Acad Sci USA, 92(16):7297-7301, 1995.
Ogris, M., Wagner, E., Tumor-targeted gene transfer with DNA polyplexes. Somat Cell Mol Genet 27(1-6): 85-95, 2002.
Plank C., Zatloukal K., Cotten M., Mechtler K., Wagner E., Gene transfer into hepatocytes using asialoglycoprotein receptor mediated endocytosis of DNA complexed with an artificial tetra-antennary galactose ligand. Bioconjug Chem, 3(6):533-539, 1992.
Carlisle, R. C., Bettinger, T., Ogris, M., Smart, S., Mautner, V., Seymour, L. W., Adenovirus hexon protein enhances nuclear delivery and increases transgene expression of polyethylenimine/plasmid DNA vectors. Mol Ther, 4(5):473-483, 2001.
Corresponding Author: Conrad Coester, Department of Pharmacy, Pharmaceutical Technology and Biopharmaceutics, Ludwig-Maximilians-University, Butenandtstr. 5-13, Munich, Germany. conrad.coester@cup.uni.muenchen.de
Published by the Canadian Society for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences.
www.cspscanada.org