J Pharm Pharmaceut Sci (www.cspscanada.org) 8(1):26-38, 2005
Formulation development, in vitro and in vivo evaluation of membrane controlled transdermal systems of glibenclamide.
Srinivas Mutalik, Nayanabhirama Udupa1
Manipal College of Pharmaceutical Sciences, Manipal, Karnataka State, IndiaReceived 5 November 2004, Revised 12 December 2004, Accepted 15 December 2004, Published 21 January 2005
PDF Version
Abstract
PURPOSE. The objective of the present study was to develop the membrane controlled transdermal systems of glibenclamide and to evaluate with respect o various in vitro and in vivo parameters. METHODS: The membrane moderated transdermal systems were prepared using drug containing carbopol gel as reservoir and ethyl cellulose, Eudragit RS-100, Eudragit RL-100 and Ethylene vinyl acetate (EVA) (2%, 9% and 19% vinyl acetate content) rate controlling membranes. The possible interaction between drug and polymer was studied by IR spectroscopy, DSC and HPTLC analysis. The formulations were subjected to various physicochemical studies, in vitro drug release studies and permeation studies through mouse skin. The blood glucose reducing hypoglycemic activity of the systems was studied in both normal and diabetic mice. Various biochemical parameters and histopathological studies were carried out after treating the diabetic mice for 6 weeks. Skin irritation tests, oral glucose tolerance test and pharmacokinetic evaluations were carried out in mice. RESULTS: The results suggested no interaction between drug and polymer. Variations in drug release/permeation profiles among the formulations containing different rate controlling membranes were observed. The scanning electron microscopic studies of EVA membranes demonstrated no changes in the surface morphology after in vitro skin permeation studies. The system with EVA rate controlling membrane (with 19% vinyl acetate) was selected for in vivo experiments. The transdermal system produced better improvement with respect to hypoglycemic activity, glucose tolerance test, all the tested biochemical, histopathological and pharmacokinetic parameters compared to oral administration, and exhibited negligible skin irritation. CONCLUSION: The present study shows that membrane controlled transdermal systems of glibenclamide exhibited better control of hyperglycemia and more effectively reversed the diabetes mellitus complications than oral glibenclamide administration in mice.
Introduction
Diabetes mellitus is a chronic metabolic disorder characterized by high blood glucose concentration-hyperglycemia-caused by insulin deficiency, often combined with insulin resistance (1). Glibenclamide, an important drug of sulfonylurea class, is currently available for treating hyperglycemia in (Non-Insulin Dependent Diabetes Mellitus (NIDDM); but has been associated with severe and sometimes fatal hypoglycemia and gastric disturbances like nausea, vomiting, heartburn, anorexia and increased appetite after oral therapy (2). Since these drugs are usually intended to be taken for a long period, patient compliance is also very important (3). We already have reported the feasibility of application of transdermal delivery for glibenclamide. Glibenclamide (molecular weight: 494 and pKa : 5.3) showed favorable partition coefficients ( log octanol/buffer: 0.32 ± 0.0.07; log isopropylmyristate/buffer: 0.50 ± 0.05) and negligible skin degradation (4, 5). In another study, we reported the formulation and evaluation of matrix type transdermal patches of glibenclamide (6). It is highly accepted that membrane controlled transdermal systems have the distinct advantage that the drug release rate, which is regulated by permeation through the rate controlling membrane, remains relatively constant as long as drug loading in the reservoir is maintained at a high level (7). Hence in the present study, we have formulated the membrane moderated transdermal systems of glibenclamide using carbopol gel as drug reservoir and various polymeric rate controlling membranes prepared by ethyl cellulose, Eudragit RL-100, Eudragit RS-100, ethylene vinyl acetate (containing 2%, 9% and 19% vinyl acetate) and evaluated with respect to various in vitro parameters (physical characteristics like thickness, drug content, moisture content/uptake, scanning electron microscopy, flatness, in-vitro release/permeation kinetics, etc) and pharmacological, biochemical and histopathological effects in vivo in mouse model.
Materials and Methods
Ethyl cellulose (EC; with an ethoxy content of 47.5-53.5% by weight and a viscosity of 14 cps in a 5% w/w, 80:20 toluene:ethanol solution at 25 ∞C) was purchased from SD Fine Chemicals Ltd., India. Eudragit RL-100 (ERL) and Eudragit RS-100 (ERS) were obtained from Rohm Pharma, Germany. Carbopol 934P NF was purchased from B.F. Goodrich, Germany. Di-n-Butylphthalate was procured from Ranbaxy Laboratories, India. Ethylene vinyl acetate (EVA) membranes with 2% vinyl acetate (VA) content (EVA2%; 3M CoTran 9726), 9% VA content (EVA9%; 3M CoTran 9702) and 19% VA content (EVA19%; 3M CoTran 9715), backing layer (a polyester film laminate; 3M Scotchpak Backing 1006) and release liner (a fluropolymer coated polyester film; 3M Scotchpak 1022 Release Liner) were gift samples from 3M Pharmaceuticals, USA. Sodium deoxycholate, anthrone, thiourea, streptozotocin, bovine serum albumin were purchased from Sigma Chemical Company, USA. Polyisobutylene was purchased from Aldrich, USA. Glibenclamide was a gift from BAL Pharma, Modi-Mundi Pharma and Wallace Pharmaceuticals, India. All the other chemicals used were of analytical/reagent grade.
Development of membrane controlled transdermal systems
The transdermal systems were fabricated by encapsulating the drug reservoir within a shallow compartment molded from a drug impermeable backing laminate and a rate controlling membrane. EC, ERL and ERS rate controlling membranes were prepared by dissolving 250, 300 and 300 mg of respective polymers in 5 ml chloroform. Di-n-Butyl phthalate (30% w/w of polymer) was used as plasticizer. The polymeric solution was poured on the mercury surface (25 cm 2 ) and dried at room temperature. After 24 h, the films were cut into 12 cm2 area. EVA rate controlling membranes were gift samples from 3M Pharmaceuticals, USA.
The reservoir (0.5% carbopol gel) of the drug was prepared as per the formula given in Table 1.
Table 1: Reservoir of the glibenclamide membrane controlled transdermal systems. The area of the transdermal system was 12 cm2 .
Carbopol was soaked in 5 ml water and neutralized using triethanolamine (q.s.) to form a gel. Drug in 5 ml ethanol was added slowly to carbopol gel with constant stirring.
Accurately weighed quantity of the gel (1 g) containing drug (12 mg of glibenclamide) was placed on a sheet of backing layer (3M Scotchpak Backing 1006) covering 3 cm x 4 cm areas. A rate controlling membrane was placed over the gel and the edges of 3 cm x 4 cm area were heat-sealed to obtain a leak proof device. To ensure intimate contact of the patch to the skin, a pressure sensitive adhesive, polyisobutylene (PIB), was applied onto rate controlling membrane (3 ml; 10% w/v in petroleum ether). A release liner (3M Scotchpak 1022 Release Liner) was placed over the adhesive coated rate controlling membrane.
Drug-polymer interaction studies
Infra-red (IR) spectroscopy (using IR-Spectrophotometer; FTIR-8300, Shimadzu, Japan; by KBr pellet method), differential scanning calorimetry (DSC) (Perkin Elmer, USA; at scanning rate of 10° C/min between 50 and 300° C), and high performance thin layer chromatographic (HPTLC) analysis (using CAMAG-HPTLC system, Switzerland) were carried out on pure substances and their physical mixtures to search the possible interaction between glibenclamide and carbopol (6).
In vitro evaluation of transdermal systems
For drug content determination, the whole contents of transdermal systems (n=3) were taken into a 100 ml volumetric flask and dissolved in methanol. The solution was filtered through 0.45- mmembrane (Nulge Nunc, UK) prior to drug analysis. The viscosity of drug containing carbopol gel was determined using Brookefield synchro electric viscometer (Brookefield Engineering Ltd., USA). The TD bar spindle of LV-4 at 12 - gear was employed.
The in vitro drug release studies and in vitro skin permeation studies were carried out using USP basket type dissolution apparatus (using 900 ml of phosphate buffer pH 7.4 as dissolution medium) and vertical type diffusion cells (using hairless mouse skin as membrane barrier), respectively (6). The morphology of the EVA rate controlling membranes before and after in vitro skin permeation experiments was analyzed by scanning electron microscopy (JEOL-JSM-840A, Japan).
In vivo studies
The animals used for in vivo experiments were adult Swiss albino mice (6-8 weeks old) of either sex, weighing 25-30 g, from the Department of Radiobiology, Kasturba Medical College, Manipal. The animals were housed in polypropylene cages, 4 per cage, with free access to standard laboratory diet (Lipton Feed, Mumbai, India) and water. They were kept at 25±1°C and 45-55% relative humidity with a 12 h light/dark cycle. The in vivo experimental protocol was approved by the Institutional Animal Ethical Committee, Kasturba Medical College, Manipal.
Hypoglycemic activity in normal mice
The hair on the backside of the mice was removed with an electric hair clipper on the previous day of the experiment. Following an overnight fast, mice were divided into 3 groups (n=6). The mice were treated as following:
Group I (Control) - 0.2 ml of 0.5% w/v sodium carboxymethyl cellulose (CMC); p.o.
Group II - Glibenclamide (5 mg/kg; p.o.). The oral doses were given using a round tipped stainless steel needle attached to 1 ml syringe and the dose of 5 mg/kg was selected by conducting a series of experiments with graded doses ranging between 1 to 10 mg/kg.
Group III - Applied with 2.5 cm2 transdermal system prepared with EVA19% rate controlling membrane, containing 2.5 mg of drug in 0.5% carbopol gel.
At time intervals between 2-24 h after treatment (acute study), blood was collected from orbital sinuses; blood glucose levels were determined using Accutrend Alpha Glucometer (Roche Diagnostics, Germany). In the long-term study, the above treatments were administered/applied once daily for 6 weeks. Blood glucose levels were determined once in every 2 weeks in over night fasted mice, 2 h after drug treatment as previously described.
Induction of diabetes mellitus and hypoglycemic activity in diabetic mice
The overnight fasted mice were made diabetic by a single intraperitoneal injection of streptozotocin (150 mg/kg; i.p.) dissolved in citrate buffer (3 mM; pH 4.5) (6). Seven days later, mice with blood glucose levels between 300-400 mg/dL were selected. The acute and long-term hypoglycemic activity of the transdermal patches was evaluated in overnight fasted diabetic mice as described in above.
Effect on glucose tolerance
After an overnight fast, mice were divided into 3 groups (n=6). Control group was administered with 0.2 ml of CMC. Other 2 groups were administered with glibenclamide (5 mg/kg; p.o.) or applied with transdermal system as described in earlier experiments. Two hours later, glucose was administered orally (2 g/kg) to all the 3 groups. Blood samples were collected just prior to and at 0.5, 1.0 and 2.0 h after the glucose feeding and glucose level was determined. The percentage change in blood glucose was estimated in comparison with the control group. During the testing period, food was not provided; but water was given ad libitum.
Biochemical and histopathological evaluation
At the end of the long-term treatment in the diabetic mice, lipid profile (high-density lipoprotein-cholesterol, triglycerides and total cholesterol), alanine transaminase (ALT), aspertate transaminase (AST), urea and creatinine levels were estimated in serum using Auto-analyzer (Hitachi 911, Japan). Then the animals were sacrificed and a part of the liver was processed for glycogen estimation and total protein (6). Pieces of liver, pancreas and stomach were subjected to histopathological studies using haematoxylene and eosin (H & E) staining.
Skin irritation test (visual and histopathological evaluation of skin)
The mice were divided into 5 groups (n=6). On the previous day of the experiment, the hair on the backside area of mice was removed. The animals of group I was served as normal, without any treatment. One group of animals (Group II, control) was applied with marketed adhesive tape (official adhesive tape in USP). Transdermal systems (blank, without drug and drug loaded) were applied onto nude skin of animals of III and IV groups. A 0.8% v/v aqueous solution of formalin was applied as a standard irritant (Group V). The animals were applied with new patch/formalin solution each day upto 7 days and finally the application sites were graded according to a visual scoring scale, always by the same investigator. The erythema scale was as follows: 0, none; 1, slight; 2, well defined; 3, moderate; and 4, scar formation. The edema scale was: 0, none; 1, slight; 2, well defined; 3, moderate; and 4, severe. After visual evaluation of skin irritation, the animals were sacrificed and skin samples were processed for histological examination (6).
Pharmacokinetic evaluation
Overnight fasted mice, whose hair was previously removed, were divided into 2 groups (n=6) and treated as follows.
Group I - Glibenclamide (5 mg/kg; p.o.).
Group II - Applied with 2.5 cm2 transdermal system prepared with EVA19% rate controlling membrane, containing 2.5 mg of drug in 0.5% carbopol gel.
Blood samples were withdrawn at different time intervals from orbital sinuses using heparinized capillaries. Plasma was separated by centrifugation using Biofuge-13 (Heraeus Instruments, Germany) and stored in vials at -70°C until further analysis.
Analysis of glibenclamide
Glibenclamide was estimated by an earlier reported reverse phase HPLC method (27). A Shimadzu Class VP series HPLC system with two LC-10AT pumps, a SPD-10A variable wavelength programmable UV/Vis detector, a SCL-10A system controller and a RP C-18 column (Luna, Phenomenex, USA; 250 mm x 4.6 mm; particle size 5 m) was used.
The system was equipped with Class VP series version 6.12 software.
Chromatographic conditions: The mobile phase consisted of 20 mM monobasic potassium dihydrogen orthophosphate in water, which was adjusted to pH 3.5 with phosphoric acid, and acetonitrile in the proportion of 60:40 v/v. The mobile phase was filtered through 0.22 m membrane filter (Sartorius, Germany). The flow rate was 1 ml/min and the column effluent was monitored at 225 nm. The total run time of the method was set at 20 min. The peaks were well resolved and the retention time for glibenclamide and glipizide (internal standard) was 9.60 and 5.96 min respectively. No interfering peaks were observed at the retention time of glibenclamide and glipizide.
Standard Solutions: A standard stock solution of glibenclamide (100 mg/ml) was prepared in acetonitrile. The calibration curve standard solutions were prepared by adding known amount of glibenclamide (concentrations: 1-20 mg/ml) and glipizide, an internal standard (1 mg/ml), to blank plasma.
Extraction procedure: A volume of 0.1 ml of blank mouse plasma and 0.1 ml of 0.1 N hydrochloric acid were mixed thoroughly. The plasma was spiked with standard glibenclamide and glipizide solutions to yield concentrations of 1-20 mg/ml of glibenclamide and 1 mg/ml of glipizide, respectively. Then the mixture was gently shaken for 3 min and then it was added with 5 ml of benzene in a 20 ml glass tube. The tube was gently shaken using cyclomixer (Remi cyclomixer, Mumbai) for 5 min and centrifuged (Remi Centrifuge, Mumbai, India) for 10 min at 3000 rpm. After centrifugation, the organic phase was transferred into a conical tube for evaporation to dryness under nitrogen. The residue was dissolved in 0.1 ml of equilibrated mobile phase by vortexing. An aliquot of 20 ml was injected into the chromatograph.
Calibration curve: Calibration curve was obtained by plotting peak area ratios of glibenclamide to glipizide (y-axis) against glibenclamide concentration (x-axis).
The pharmacokinetic parameters were calculated using noncompartmental pharmacokinetics data analysis software, PK Solutions 2.0Ô.
Statistical analysis
The results were analyzed by Student's t-test using Graph Pad Instat Software (Version: 1.13). Difference below the probability level 0.05 was considered statistically significant.
Results
Drug-polymer interaction studies
The IR spectral analysis of glibenclamide alone showed that, the principal peaks were observed at wave numbers of 1527.50, 1157.2, 1618.2, 1714.6 and 819.7 confirming the purity of the drug as per established standards (8). In the IR spectra of the physical mixture of glibenclamide and carbopol, the major peaks of glibenclamide were 1527.50, 1157.2, 1618.2, 1716.5 and 819.7 wave numbers. However, some additional peaks were observed with physical mixtures, which could be due to the presence of polymer.
The DSC analysis (Figure 1) of pure glibenclamide showed a sharp endotherm peak at 175.16°C corresponding to its melting point.
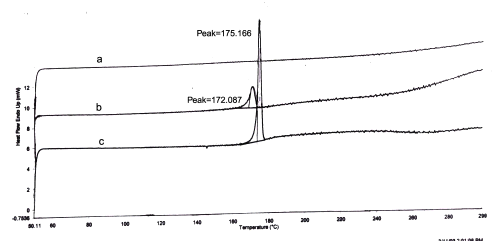
Figure 1: DSC Thermograms. a=Carbopol; b=mixture of Carbopol and glibenclamide; c=glibenclamide.
The DSC analysis of physical mixture of drug and carbopol revealed negligible change in the melting point of glibenclamide in the presence carbopol (172.08°C for the mixture of glibenclamide and carbopol). In HPTLC analysis, the Rf value of pure glibenclamide was found to be 0.92. In the presence of polymer, the Rf value of the drug was unchanged and found to be 0.92.
Drug content and viscosity
The drug content of the transdermal systems was ranged between 99.95±0.12 and 99.04±0.13%. The viscosity of reservoir of membrane controlled transdermal systems (0.5% carbopol gel containing glibenclamide) was uniform among the batches and found to be 17100±150 cps.
In vitro drug release studies
The results of in vitro drug release studies from transdermal systems are depicted in Figure 2.
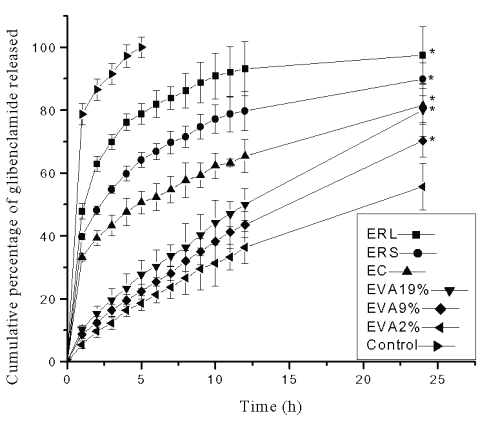
Figure 2: Cumulative percentage of glibenclamide released in in vitro dissolution studies from reservoir transdermal systems prepared using ethyl cellulose (EC), Eudragit RL-100 (ERL), Eudragit RS-100 (ERS) and ethylene vinyl acetate (EVA) copolymer rate controlling membranes containing 2%, 9% and 19% VA (EVA2%, EVA9% and EVA19% respectively). Each point represents Mean±SE, n=3; * significant compared to EVA2%; Control=Suspension of glibenclamide in phosphate buffer.
The formulations with ERL rate controlling membrane exhibited the greatest cumulative percentage of drug release value (97.45±9.01%) followed by ERS (89.85±5.25%), EC (81.55±5.32 %), EVA19% (80.02±8.32%), EVA9% (70.25±5.24%) and EVA2% (55.65±7.36%) membrane containing systems at the end of 24 h.
In vitro skin permeation studies
The results of in vitro skin permeation of glibenclamide from patches are shown in Figure 3.
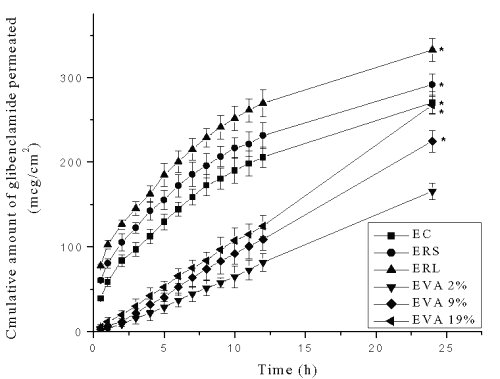
Figure 3: Cumulative amount of glibenclamide permeated (mg/cm2) across mouse skin from reservoir transdermal systems prepared using ethyl cellulose (EC), Eudragit RL-100 (ERL), Eudragit RS-100 (ERS) and ethylene vinyl acetate (EVA) copolymer rate controlling membranes containing 2%, 9% and 19% VA (EVA2%, EVA9% and EVA19% respectively). Each point represents Mean±SE, n=3; * significant compared to EVA2%.
The formulations (1 cm2 ) with ERL rate controlling membrane exhibited the greatest (332.54±13.36 mg) cumulative amounts of drug permeation followed by ERS (291.57±12.31 mg), EC (270.23±12.62 mg), EVA19% (267.25±10.28 mg), EVA9% (224.58±12.61mg) and EVA2% (165.58±9.65 mg) membrane containing devices at the end of 24 h. The transdermal systems with EVA2% rate controlling membrane showed significantly low (p<0.05) cumulative amount of drug permeation compared to other rate controlling membrane systems.
Scanning electron microscopy
Figure 4 shows the microstructure of different EVA rate controlling membranes of reservoir systems before and after the in vitro drug permeation experiments.

Figure 4: SEM photographs of EVA rate controlling membranes A and A1= EVA2% rate controlling membranes before and after in vitro skin permeation studies, respectively. B and B1= EVA9% rate controlling membranes before and after in vitro skin permeation studies, respectively. C and C1= EVA19% rate controlling membranes before and after in vitro skin permeation studies, respectively.
No considerable difference was observed in the microstructure of EVA films before and after in vitro permeation experiments.
In vivo studies
Acute hypoglycemic activity
The results of acute hypoglycemic activity of transdermal system in comparison with glibenclamide (5 mg/kg; p.o.) in both normal and diabetic mice are shown in Table 2.
Table 2: Reduction in blood glucose levels after oral and transdermal administration of glibenclamide in normal and diabetic mice (acute study). All values are expressed as Mean±SE, n=6; CMC=Carboxymethyl cellulose; TP-R=Reservoir transdermal system; EVA=Ethylene vinyl acetate; GLB=Glibenclamide; * significant compared to control (p<0.05); # significant compared to GLB (p<0.05); ♠ significant compared to DC (Diabetic control) (p<0.05); ♣ significant compared to GLB (p<0.05).
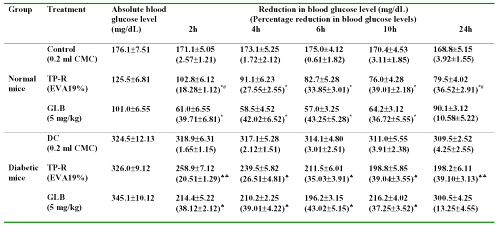
The blood glucose reducing effect was significant in oral and transdermal patch treated animal groups upto 10 h, compared with control group (p<0.05). Glibenclamide (oral) produced a decrease of 39.71±6.81 (normal mice, p<0.05 compared to control) and 38.12±2.12% (diabetic mice, p<0.05 compared to diabetic control) in blood glucose levels at 2 h. In case of transdermal system, the blood glucose reducing response was gradual. A maximum blood glucose reducing response was observed after 6 h and thereafter remained stable upto 24 h.
In orally glibenclamide treated group, the blood glucose levels decreased after 6 h. The blood glucose levels at the end of 24 h were only 10.58±5.22% and 13.25±4.55% in normal and diabetic mice, respectively. On the other hand, the transdermal system produced significant reduction in blood glucose levels upto 24 h compared to control (p<0.05). The untreated group did not show any noticeable hypoglycemia.
Long-term hypoglycemic activity
The results of long-term hypoglycemic activity of transdermal system in comparison with oral glibenclamide in both normal and diabetic mice are shown in Table 3.
Table 3: Absolute blood glucose levels after oral and transdermal administration of glibenclamide in long-term study. All values are expressed as Mean±SE, n=6; CMC=Carboxymethyl cellulose; TP-R=Reservoir transdermal system; EVA=Ethylene vinyl acetate; GLB=Glibenclamide; * significant compared to control (p<0.05); # significant compared to GLB (p<0.05); ♠ significant compared to DC (Diabetic control) (p<0.05); ♣ significant compared to GLB (p<0.05).
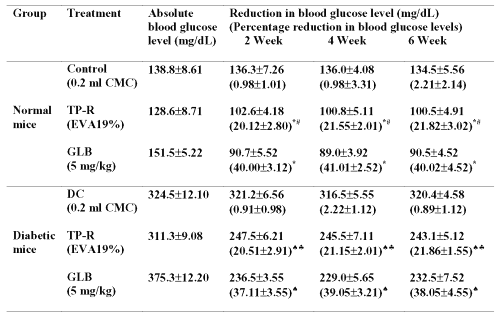
The transdermal system produced significant (p<0.05) blood glucose reducing effect upto 6 weeks without causing severe hypoglycemia in the initial hours of treatment, which was observed with oral administration.
Effect on glucose tolerance (GTT)
The control group showed high-elevated blood glucose levels (p<0.05) after glucose administration (+81.04±2.81, +62.05±3.41 and +15.01±4.01% at 0.5, 1.0 and 2.0 h, respectively) (Figure 5).
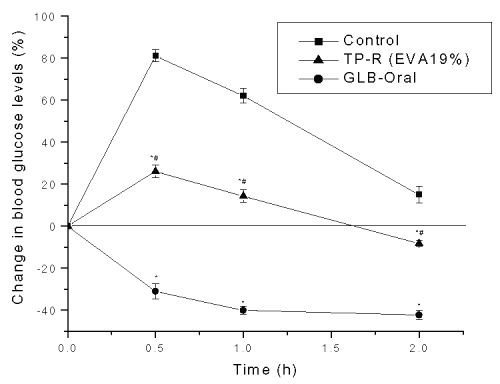
Figure 5: Effect on glucose tolerance after oral and transdermal administration of glibenclamide in mice. Each point represents Mean±SE, n=6; * significant compared to control (p<0.05); # significant compared to GLB (p<0.05).
The hypoglycemia produced after transdermal delivery was significantly (p<0.05) lower than the control group. On the contrary, the orally glibenclamide administered group showed severe hypoglycemia ranging from -30.95±3.52 to -42.20±2.22% at all intervals of the study period.
Biochemical and histopathological evaluation
The results of biochemical studies are shown in Table 4.
Table 4: Liver protein and glycogen levels and serum lipid profile (TC, TG and HDL-C), alanine transaminase, aspertate transaminase, urea and creatinine levels in diabetic mice after oral and transdermal administration of glibenclamide. All values are expressed as Mean±SE, n=6; NC=Normal control; DC=Diabetic control; CMC=Carboxymethyl cellulose; TP-R=Reservoir transdermal system; EVA=Ethylene vinyl acetate; GLB=Glibenclamide; TC=Total cholesterol; TG=Triglycerides, HDL-C= High density lipoprotein-cholesterol, ALT=Alanine transaminase, AST=Aspertate transaminse; # significant compared to NC (p<0.05); *significant compared to DC (p<0.05).
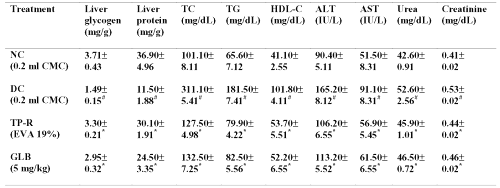
The glycogen and total protein levels in the liver of diabetic mice were significantly lowered compared to normal mice (p<0.05). The oral as well as transdermal treatment of glibenclamide significantly (p<0.05) increased liver glycogen and total protein levels at the end of 6 weeks. The serum lipid profile (total cholesterol, triglycerides and high-density lipoprotein-cholesterol), hepatic enzymes (ALT and AST), urea and creatinine levels were significantly increased in diabetic control mice compared to normal mice (p<0.05). The glibenclamide treatment (both oral and transdermal) significantly (p<0.05) reversed these changes at the end of 6 weeks.
The histopathological studies of liver, pancreas and stomach from diabetic mice are presented in Table 5.
Table 5: Histopathological evaluation of liver, pancreas and stomach from diabetic mice treated with oral and transdermal administration of glibenclamide. CA=Cellular atypia; Deg=Degeneration; Nec=Necrosis; Con=Congestion; Inf=Inflammation; GLB=glibenclamide; TP-R=Reservoir transdermal system; EVA=Ethylene vinyl acetate. Histopathological scale: + = slight; ++ = moderate; +++ = severe.
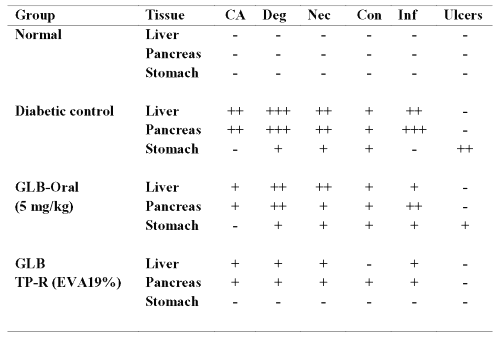
The liver and pancreas from untreated diabetic mouse showed severe/moderate cellular atypia, inflammation, necrosis, degeneration and congestion. The stomach samples from diabetic mice showed moderate ulceration. The severe/moderate toxic manifestations including gastric ulceration were considerably reversed with oral but especially with transdermal administration of glibenclamide by controlling the hyperglycemia.
Skin irritation test
The results (Table 6) showed that the prepared systems (both blank and drug loaded) and USP adhesive tape produced negligible erythema and edema.
Table 6: Results of skin irritation test. Visual observation values are expressed as Mean±SE, n=6; * significant compared to formalin (p<0.05); GLB=Glibenclamide; TP-R=Reservoir transdermal system; EVA=Ethylene vinyl acetate; Blank=Without drug; Inf= Inflammation. Erythema scale: 0, none; 1, slight; 2, well defined; 3, moderate; and 4, scar formation. Edema scale: 0, none; 1, slight; 2, well defined; 3, moderate; and 4, severe. Histopathological scale: + = slight; ++ = moderate; +++ = severe.
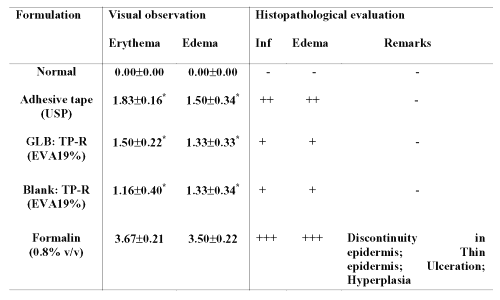
On the other hand, standard irritant, formalin produced severe erythema and edema. The histopathological examination of the skin indicated that adhesive tape and prepared patches produced mild inflammation and edema. Formalin produced high grade of irritation, indicated by `severe' inflammation and edema besides showing discontinuity in epidermis, thin epidermis, ulceration and hyperplasia.
Pharmacokinetic studies
The plasma concentrations of glibenclamide after transdermal and oral administration against time are shown in Figure 6.
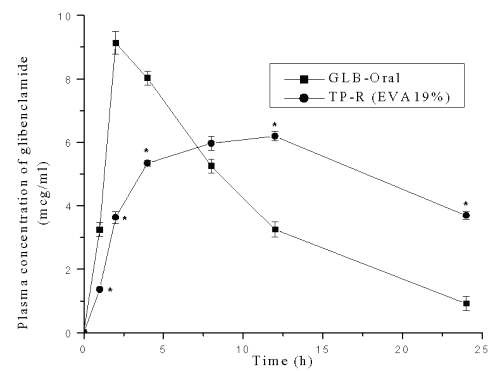
Figure 6: Plasma concentration-time profile of glibenclamide after oral and transdermal system treatment in mice. * Significant compared to GLB-Oral (p<0.05). Each point represents Mean±SE; n=6.
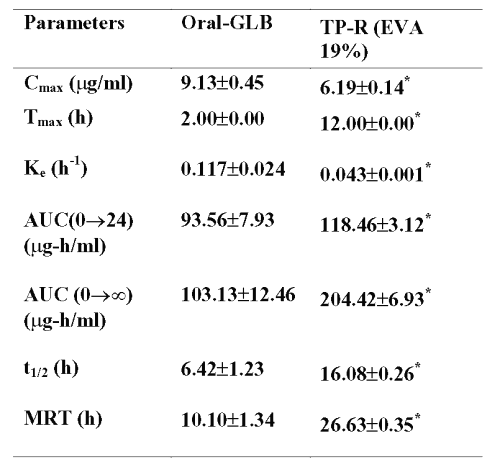
Peak plasma concentration, Cmax , after oral administration was 9.13±0.45 mg/ml and tmax was 2.0 h. In the case of transdermal system, the Cmax and tmax were 6.19±0.14 mg/ml and 12.0 h respectively. The pharmacokinetic parameters were calculated from the plasma concentrations of the drug and recorded in Table 7.
Table 7: Pharmacokinetic parameters of glibenclamide after oral and transdermal administration All values are expressed as Mean±SE, n=6; GLB=Glibenclamide; TP-R=Reservoir transdermal system; EVA=Ethylene vinyl acetate; Cmax=Maximum concentration; Tmax=Time of maximum concentration; Ke=Elimination rate constant; AUC=Area under plasma concentration-time curve; t1/2=Elimination half-life; MRT=Mean residential time. * significant compared to oral GLB (p<0.05).
Discussion
In this study, membrane moderated transdermal systems containing glibenclamide were prepared using different rate controlling membranes. It was desired to develop a transdermal system that allows one to provide an optimum drug release via the most appropriate choice of rate controlling membrane and finally to produce the desired overall constant/controlled drug release. Ethanol (50% w/w) was incorporated in the reservoir of the transdermal system as it significantly enhanced the permeation rate of glibenclamide in our earlier study (4, 5).
The pharmacokinetic parameters obtained with glibenclamide transdermal system were significantly different (p<0.05) from those obtained with respective oral glibenclamide administration, which could be due to the rapid absorption of drugs via oral route; whereas drug in transdermal route were slowly but continuously absorbed. With respect to all in vivo experiments, similar results were observed with matrix transdermal patches in our earlier study.
In drug-carbopol interaction studies, no distinct difference in the IR peaks and melting point (DSC analysis) of drug in the physical mixture and Rf values of drug (HPTLC analysis) in the polymeric solution used in our study indicates that the carbopol do not alter the performance characteristics of the drug from the systems studied. All these results suggest that there is no interaction between glibenclamide and carbopol. Good uniformity with respect to drug content and viscosity among the batches with all formulations was observed.
In the membrane controlled transdermal systems, drug reservoir is encapsulated in a shallow compartment molded from a drug impermeable backing membrane while the drug delivery side is covered by rate controlling polymeric membrane. These systems have the advantage that the drug release rate, which is controlled by the rate controlling membrane, remains relatively constant as long as drug loading in the reservoir is maintained at a high level and thus provide a zero order drug release (7). But in the in vitro dissolution studies, the systems with ERL, ERS and EC membranes did not provide a constant drug release rate and showed high release of drug in the initial hours. This may be due to the loss of integrity of the films by means of solubilization of a part of the polymer by ethanol, which was incorporated to the drug reservoir to enhance the drug permeation rate. In addition, direct exposure of Eudragit films to the dissolution medium might also be responsible for the initial burst release as they are permeable to aqueous medium (9).
The cumulative amount of drug released at the end of 24 h was depending on the hydrophilicity of the polymers (ERL>ERS>EC). ERL films tend to swell more than ERS films in aqueous medium due to the higher concentration of hydrophilic quaternary groups. Also, ERL membranes are more permeable to aqueous medium than other two membranes (10). On the other hand, the systems with EVA membranes exhibited a constant drug release rate upto 24 h as the integrity of these membranes was unaffected by either ethanol content of the drug reservoir or direct contact with aqueous medium. The release rate studies revealed that, as the vinyl acetate content in copolymer increases, the cumulative percentage of drug release also increases, i.e., the membrane shows a lower resistance to the permeation of drug molecules. These observations are in accordance with the earlier findings (11).
In the in vitro skin permeation studies also, the systems with ERL, ERS and EC membranes did not provide a constant drug release and showed high permeation of drug in the initial hours. This is again because of the ethanol content (50%) of the drug reservoir, which might have partly dissolved the polymeric membranes and thereby disrupting the integrity of the membrane (9). The ERL, ERS and EC membranes showed loss of uniform structure after fabrication into final system. This was confirmed by examining the films after 24h of fabrication of final systems where the ethanol containing gel is in direct contact with membrane or after in vitro skin permeation experiments. It was also supported by the reduced flatness of these rate controlling membranes after preparing the final transdermal devices. The films prior to fabrication into final systems exhibited 100% flatness indicating no constriction. After 24 h of fabrication, they showed reduced flatness values (80%, 80% and 90% of flatness for ERL, ERS and EC membranes, respectively). These observations suggest that the films prepared by ERL, ERS and EC lose their integral structure after coming into contact with drug reservoir.
Hence it was thought to use those membranes, which retain their integrity when they come in contact with the drug reservoir. The membranes prepared by ethylene vinyl acetate copolymer (EVA) are widely used to control the rate of drug release from many transdermal drug delivery systems (11, 12). These membranes did not lose their integral structure when fabricated into final system and also after in vitro skin permeation studies. This could be due to the ability of these films to resist the effect of ethanol in drug reservoir. This is also supported by the SEM studies of the EVA films before fabricating into final system and after in vitro skin permeation experiments, where the films maintained uniform and smooth surface, 100% flatness and integrity of the structure after permeation experiments. Therefore EVA rate controlling membranes are suitable for reservoir transdermal systems in the present study.
The cumulative amount of drug permeated at the end of 24 h was increased as the vinyl acetate (VA) content in the EVA rate controlling membranes was increased. This observation is in accordance with the earlier reports where drug permeation was increased as the vinyl acetate content in the EVA copolymer was increased (11,13,14). It is well recognized that it is possible to alter the permeability of EVA copolymer membranes by varying vinyl acetate content. The changes in the permeability have been attributed to the alterations in the glass transition temperature and crystallinity of the polymer. As the vinyl acetate content increases, the crystallinity of the polymer decreases rapidly and this could be the reason for high permeation rate observed with EVA membrane with 19% VA content in comparison with EVA membranes containing 9% and 2% VA in the present study (15).
The cumulative amounts of glibenclamide permeated per square centimeter of transdermal devices with ERL, ERS and EC rate controlling membranes through the mouse skin when plotted against time, the permeation profiles of drug seem to follow mixed order/apparent zero order kinetics as observed with matrix systems (Figure 3). High amount of drug was released in the initial hours because of increased permeability of the membranes by ethanol. However, later the drug was released depending on the concentration and thus altering the system towards a first order pattern.
The in vitro permeation profiles of formulations with ERL, ERS and EC membranes did not fit into zero order behavior completely and they could be best expressed by Higuchi's equation (R2= 0.9976 to 0.9990) (16, 17).
The data was further treated as per the following equation (18,19).
Mt /M¥ =K.t n ,
where, Mt /M¥ is the fractional release of drug, Mt is the amount released at time t, M¥ is the total amount of drug contained in the transdermal patch, t is the release time, K is a kinetic constant and n is the diffusional release exponent indicative of the operating release mechanism. The Fickian diffusion dominated drug release from these systems was further confirmed by the n values (0.3897 £ n ³ 0.4934) obtained by the plots of Korsmeyer's equation. These observations demonstrate that, if the drug release is not properly regulated by the rate controlling membranes, the system may exhibit diffusion predominated drug release instead of zero order kinetics.
The reservoir systems with EVA rate controlling membranes exhibited a zero order pattern throughout 24 h of the permeation studies (R2 =0.9922 to 0.9995) as the integrity of these membranes was not affected by the drug reservoir (Figure 3). Since the concentration of the drug in equilibrium with the inner surface of the enclosing membrane is constant, zero order permeation profile is generally expected with membrane controlled systems (20). These results are in agreement with earlier reports where membrane controlled transdermal systems with EVA rate controlling membranes showed zero order release (11,20).
The systems with ERL, ERS and EC rate controlling membranes exhibited high drug release in the initial hours and on the whole, the drug permeation pattern was diffusion dominated. The membranes lost their integrity after formulating into final systems. Hence, further systems were developed using EVA rate controlling membranes having varying VA content (EVA2%, EVA9% and EVA19%). Among these systems, the device with EVA19% membrane exhibited high cumulative amounts of drug permeation at the end of 24 h. The release/permeation rate was in a constant manner throughout 24 h. Thus this system was selected for further in vivo studies.
The results of short-term blood glucose reducing effect clearly show that the system could sustain the drug release for a period of 24 h when compared with oral administration where the effect declined after 6 h in agreement with short half-life of glibenclamide (21). Also the study clearly shows that severe hypoglycemia associated with oral administration of glibenclamide can be successfully overcome by membrane moderated transdermal system. The results of long-term study indicated that transdermal system provides optimum blood glucose reduction upon chronic application without producing drastic decrease in blood glucose levels in initial time intervals. The results of GTT show that the glucose tolerance curve was completely inhibited in the treated groups. Transdermal route effectively maintained the normoglycemic levels in contrast to the oral group, which produced remarkable hypoglycemia, an indication that a similar incident might be prevented in diabetic patients.
In biochemical and histopathological studies, the favourable results from glibenclamide treatment with respect to liver glycogen, liver protein, serum lipid profile, serum urea and serum creatinine levels could be due to increased insulin release and peripheral uptake of glucose after drug treatment, as explained before6. The membrane moderated systems of glibenclamide also produced improved repair of the tissues after diabetes induced tissue injury in comparison with oral administration, which could be due to better control of hyperglycemia. The transdermal systems (both drug loaded and blank) did not produce any cutaneous reaction in skin irritation test indicating they are well tolerated by the subjects. Similar results have been reported by Krishna and Pandit (20) and Kulkarni et al (22). In the pharmacokinetic study, though the rise in drug concentration was slower than oral administration, the drug concentration in plasma remained high for longer period with transdermal systems. Glibenclamide binds to plasma proteins to the extent of 99% (6). The prolongation of plasma half life by transdermal systems indicate that the drug when administered by transdermal systems will remain in the body for a longer period and thus will exert a sustained action. The significantly less elimination rate constants (Ke) and high mean residential time (MRT) values of glibenclamide obtained with transdermal systems further support the sustained or slow release of drug from the transdermal systems. Although, the Cmax was significantly less with transdermal devices, the AUC values were significantly high compared to oral route, which could be due to maintenance of concentration of drug with in the pharmacologically effective range for longer period of time from the transdermal systems. The significantly high AUC values observed with transdermal devices also indicate increased bioavailability of drug from these systems compared to oral administration.
In the present study, with respect to the in vivo studies conducted, membrane moderated transdermal system of glibenclamide produced better improvement with all the tested parameters compared to oral administration. This could be due to slow and continuous supply of glibenclamide at a desirable rate to systemic circulation by transdermal patch, which improved day-to-day glycemic control in diabetic subjects (23). Further, the slow and sustained release of the drug from the transdermal systems might reduce manifestations like sulfonylurea receptor down regulation and the risk of chronic hyper-insulinemia, a major risk factor for atherosclerosis frequently associated with oral therapy of glibenclamide (2,24,25). The present study shows that membrane moderated transdermal system of glibenclamide exhibited better control of hyperglycemia besides more effectively reversing the complications associated with diabetes mellitus than oral glibenclamide administration in mice.
In one of our earlier studies, we calculated the target permeation rate for transdermal delivery of glibenclamide in man (60 kg) as 193.8 mg/h based on available pharmacokinetic data (4). In this study, the cumulative amount of drug permeated from the system (1 cm2 ) at the end of 24 h is 267.25±10.28 mg. Hence the transdermal system with an area of about 18 cm2 would be sufficient to provide an optimum effect. But, it is well known that human skin is less permeable compared to mouse skin (26). However, in the view of encouraging results obtained in mice, it can be predicted that the required minimum effective concentration could be achieved within an appreciable range of application area in humans in spite of the greater barrier properties of human skin when compared to mouse skin.
Acknowledgements
We are thankful to Council for Scientific and Industrial Research (CSIR), India for providing Senior Research Fellowship to one of the authors (Dr. Srinivas Mutalik). We are thankful to Ms. Chetana, Ms. Sulochana and Mr. Subramanian for help and cooperation, Dr. G.C. Jagetia (Head, Department of Radiobiology, KMC, Manipal) and Dr. P. Uma Devi (Head, Department of Research, J. N. Cancer Hospital, Bhopal) for providing some laboratory facilities, and Dr. Sharath Kumar, Pathologist, District Hospital, Udupi for helping in histopathological studies. We are indebted to Bal Pharma, Modi-Mundi Pharma and Wallace Pharmaceuticals, India for providing glibenclamide as a gift sample.
References
Nolte, M.S., Karam, J.H., Pancreatic hormones and antidiabetic drugs, in Katzung BG (eds), Basic and clinical pharmacology. 8th ed., Lange Medical Books/ McGraw-Hill Publishing Division, New York, pp 711-734, 2001
Davis, S.N., Granner D.K., Insulin, oral hypoglycemic agents, and the pharmacotherapy of the endocrine pancreas, in Hardman JG: Limbird LE (eds), The pharmacological basis of therapeutics. 9th ed., McGraw-Hill Co., New York, pp 1487-1517, 1996
Takahshi, Y., Furuya, K., Iwata, M., Onishi, H., Machida, Y. and Shirotake, S., Trial for transdermal administration of sulfonylureas. Yakugaku Zassi, 12: 1022-1027, 1997
Mutalik, S and Udupa, N., Transdermal delivery of glibenclamide and glipizide: In vitro permeation studies through mouse skin. Pharmazie, 12: 838-841, 2002
Mutalik, S and Udupa, N., Effect of some penetration enhancers on the permeation of glibenclamide and glipizide through mouse skin. Pharmazie, 12: 891-894, 2003
Mutalik, S and Udupa, N., Glibenclamide transdermal patches: Physicochemical, pharmacodynamic, and pharmacokinetics evaluations. J Pharm Sci, : 1577-1594, 2004
Guy, R.H, Hadgraft, J., Selection of dug candidates for transdermal drug delivery, in Guy RH: Hadgraft J (eds), Transdermal drug delivery Developmental Issues and Research Initiatives, Marcel Dekker, New York, pp 59-81, 1989
Moffat, A.C., Clarke's Isolation and Identification of Drugs, 2nd ed., The Pharmaceutical Press, London, 1986
Kibbe, H.A., Hand Book of Pharmaceutical Excipients, 3rd ed., American Pharmaceutical Association and The Pharmaceutical Press, London, 2000.
Devi, K., Saisivam, S., Maria, G.R. and Deepti, P.U., Design and evaluation of matrix diffusion controlled transdermal patches of verapamil hydrochloride. Drug Dev Ind Pharm, 5: 495-503, 2003.
Ocak. F. and Agabeyoglu, I., Development of a membrane controlled transdermal therapeutic system containing isosorbide dinitrate. Int J Pharm, 180: 177-183, 1991.
Friend, D.R., Catz, P., Heller, J. and Okagaki, M., Transdermal delivery of levonorgestrel. V. Preparation of devices and evaluation in vitro. Pharm Res, 11: 938-944, 1989
Kagayama, A., Mustafa, R., Akoho, E., Khawam, N., Truelove, J. and Hussain, A., Mechanism of diffusion of compounds through ethylene vinyl acetate copolymers I. Kinetics of diffusion of 1-chloro-4-nitrobenzene, 3-4-dimethylphenol and 4-hexylresorcinol. Int J Pharm, 18: 247-258, 1984
Peterson, T., Burton, S., Englund, B. and Grosh, S., In vitro permeability of poly(ethylene vinyl acetate) and microporous polyethylene membranes. Proc Int Symp Control Release Bioactive Mater, 17: 411-412, 1990
Baker, R.W., Heller, J., Materials selection for transdermal drug delivery systems, in Guy RH: Hadgraft J (eds), Transdermal drug delivery Developmental Issues and Research Initiatives, Marcel Dekker, New York, pp 293-311, 1989.
Chien, Y.W., Transdermal therapeutic systems, in Robinson JR: Lee VHL (eds), Controlled drug delivery fundamentals and applications, 2nd ed., Marcel Dekker, New York, pp 524-552, 1987
Higuchi, T., Mechanism of sustained action medication-Theoretical analysis of rate release of solid drugs dispersed in solid matrices. J Pharm Sci, 52: 1145-1149, 1963
Korsmeyer, R.W., Gurny, R., Doelker, E., Buri, P. and Peppas, N.A., Mechanism of solute release from porous hydrophilic polymers. Int J Pharm, 15: 25-35, 1983
Guyot, M. and Fawaz, F., Design and in vitro evaluation of adhesive matrix for transdermal delivery of propranolol. Int J Pharm, 204: 171-182, 2000
Krishna, R. and Pandit, J.K., Transdermal delivery of propranolol. Drug Dev Ind Pharm, 15: 2459-2465, 1994
Christ,,O.E., Heptner, W. and Rupp, W., Pharmacokinetics of a new highly effective sulfonylurea derivative. Acta Diabetol Lat, 1: 101-115, 1969
Kulkarni, R.V., Mutalik, S. and Hiremath, D., Effect of plasticizers on the permeability and mechanical properties of Eudragit films for transdermal application. Indian J Pharm Sci, 1: 28-31, 2002
DCCT Research Group., The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin dependent diabetes mellitus. N Engl J Med, 329: 977-986, 1993
Faber, O.K., Beck-Neilsen, H. and Binder, C., Acute actions of sulfonylurea drugs during long-term treatment of non-insulin dependent diabetes mellitus. Diabetes Care, 13: 26-31, 1990.
Bitzen, P.O., Melander, A. and Schersten, B., Long-term effects of glipizide on insulin secretion and blood glucose control in patients with non-insulin dependent diabetes mellitus. Euro J Clin Pharmacol, 42: 77-82, 1992
Catz, P. and Friend, D.R., Transdermal delivery of levonorgestrel. VIII. Effect of enhancers on rat skin, hairless mouse skin, hairless guinea pig skin, and human skin. Int J Pharm, 58: 93-102, 1990
Emilsson, H., Sjoberg, S., Svedner, M. and Christenson, I., High performance liquid chromatographic determination of glibenclamide in human plasma and urine. J Chromatography, 383: 93-102, 1986
Corresponding Author: N. Udupa, Professor, Manipal College of Pharmaceutical Sciences, Manipal- 576 104, Karnataka, India. udupa1553@yahoo.com
Published by the Canadian Society for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences.
http://www.cspscanada.org