J Pharm Pharmaceut Sci (www.cspscanada.org) 8(1):69-75, 2005
Antinociceptive effects of some synthetic d-valerolactones.
Patrícia de Aguiar Amaral, Ana Maria Bergold, Vera Lucia Eifler-Lima1
Programa de Pós-Graduação em Ciências Farmacêuticas, Universidade Federal do Rio Grande do Sul - UFRGS, Porto Alegre/RS, BrazilEverton Melo dos Santos, Eduardo Rolim de Oliveira
Instituto de Química, UFRGS, Porto Alegre/RS, BrazilFátima Campos-Buzzi, Valdir Cechinel-Filho
Núcleo de Investigações Químico-Farmacêuticas (NIQFAR)/CCS, Universidade do Vale do Itajaí - UNIVALI, Itajaí/SC, Brazil.Received 23 November 2004, Revised 14 January 2005, Accepted 4 February 2005, Published 8 April 2005
PDF Version
Abstract
PURPOSE. A series of d-valerolactones has been synthesized in good yields, by reaction of ethyl acetoacetate with several aldehydes in presence of LDA. METHODS AND RESULTS. The in vivo analgesic activity of these compounds has been evaluated. The results indicate that the lactones synthesized showed an important antinociceptive effect at 10 mg/kg, administered intraperitoneally in mice, which significantly inhibited the abdominal constrictions induced by acetic acid when compared to acetylsalicylic acid and acetaminophen in the same dose, and increased significantly the thermal sensibility at the hot-plate method, although they were less effective than morphine in the same assay. CONCLUSIONS. The antinociceptive models employed here reveal a potential analgesic effect of the d-valerolactones synthesized. Further investigations are needed to discern which mechanism of action is concern.
Introduction
Lactones are an important class of compounds with a wide range of biological activity. Some pharmacological activities have been reported for lactones are: inhibitors of cholesterol biosynthesis (1), antifungal (2), antiviral and protease inhibitors (3), treatment of anxiety (4) and some of them have been produced antinociception in vivo in several nociceptive tests (5), and anti-inflammatory activity (5,6). In addition, lactones can serve as a building-block for combinatorial synthesis (7, 8). For these reasons, the synthesis of lactones remains of increasing therapeutic interest. The variability of biological activities of d-lactones prompted us to synthesize a series of ketolactones like 3a-e, Figure 1. In this work we focused our attention to evaluate their possible analgesic effects in mice by performing antinociceptive tests which comprise a set of pharmacological assays related to analgesic activity.
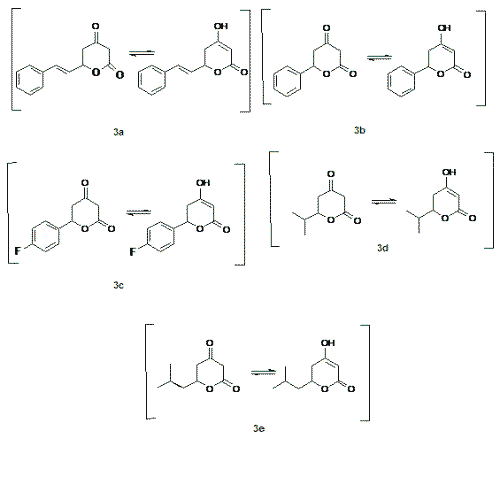
Figure 1: d-valerolactones synthesised in this work.
Results and Discussion
CHEMISTRY
Several routes are known for the preparation of different d-lactones (9-12). One of the simplest methods for the synthesis of the title compounds is to use b-ketoesters in alkaline medium in order to generate the dienolate for further aldol reaction with aldehydes. This protocol was published by Reffstrup and Boll in 1976 (11), and it is efficient for fast studies of structure-activity relationship (SAR) of d-valerolactones because it is a one-pot method and the yield and purity of products are both high.
The compounds of interest 3a-e were obtained using ethyl acetoacetate 1 as starting material in reaction with different aldehydes (2a-h) in aprotic enolate production condition, in an improvement of the method described by Reffstrup and Boll (11), as seen in Scheme 1.

Scheme 1.
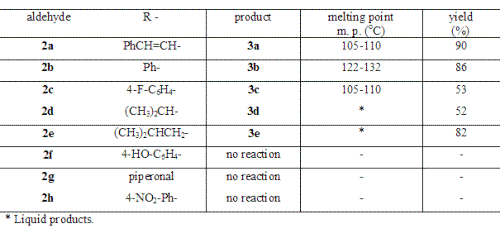
In this work, we prepared a series of 4-oxo d-valerolactones 3a-3e in the one-pot reaction. According the medium employed these lactones can present the enol form. As we can observe in Table 1, the reactions with cinnamaldehyde (2a), benzaldehyde (2b), 4-fluorobenzaldehyde (2c), isobutyraldehyde (2d) and, isovaleraldehyde (2e) lead to the corresponding d-valerolactones with good yields. On the other hand, with less activated aldehydes like 4-hydroxybenzaldehyde (2f) and piperonal (2g), the reactions, in the used conditions (THF/0°C/20 min), yielded unchanged starting material. The same results were obtained with 4-nitrobenzaldehyde (2h).
Table 1: Results from the reactions with aldehydes 2.
The lactones were efficiently prepared by addition of a more nucleophylic anion derived from ethyl acetoacetate 1 into aldehyde 2.
The NMR spectra (see experimental), with DMSO-d6 as solvent, of compounds 3a, 3b, and 3c have shown characteristic resonance signals corresponding to the enol form, caused by the hydrogen bond from DMSO-d6 and 4-hydroxy group. The 1H NMR spectra of these compounds present a singlet between δ 4.50 ppm and δ 5.11 ppm assigned to vinylic hydrogen H-3. In addition, the 13C NMR spectra of 3a-c present the signal at δ 91.0 ppm attributed to sp2 carbon C-3 and other at δ 173 ppm corresponding to quaternary C-4. On the other hand, when the solvent is CDCl 3 in 13 C NMR spectra lactones 3d and 3e present the signal at δ 200 ppm and at δ 201 ppm respectively, attributed to oxo group. These assignments are in accordance with the lactone ring obtained and with the literature (11-12). Furthermore, a doublet was observed at δ 162 ppm in a 13C NMR spectrum from lactone 3c, corresponding to an aromatic C-F coupling, with a J1CF = 244 Hz. The FT IR spectra of these lactones showed the absorption bands for carbonyl groups from 6-member lactones and ketones.
PHARMACOLOGICAL ACTIVITY
In order to select the more active compound, a preliminary trial was necessary. For this purpose, the compounds 3a, 3b, 3c and 3d were tested for their antinociceptive effect using the abdominal constrictions induced by acetic acid essay, administered intraperitoneally in Swiss mice at 10 mg/Kg, as reported previously (13-15) and as outlined in Table 2.
Table 2: Analgesic effect of d-valerolactones synthesized and of reference drugs in an acetic acid-induced abdominal constriction essay in Swiss mice at 10 mg/kg, administered intraperitoneally.

According the results, all compounds tested, 3a, 3b, 3c and 3d inhibited significantly the abdominal constrictions induced by acetic acid, causing inhibitions of 69.1%, 90.4%, 96.1% and 64.8%, respectively. The drugs used as reference, acetylsalicylic acid and acetaminophen, showed less inhibition at same dose, 35% and 38%, respectively.
The compounds 3b and 3c caused the most pronounced effect and were analyzed in detail by the same experimental model and by other chemical and thermal nociceptive assays. The aromatic ring bounded directly to C6 of the lactone cycle seems to be responsible for the effect observed on compounds 3b and 3c, when compared with the results obtained by the analogues 3a and 3d. Moreover, the only structural difference existing among the compounds 3b and 3c is the presence of a fluorine atom at para -position of the phenyl ring in 3c, which presented the best activity, suggesting that the electronegativity (inductive effect) and the hydrophobic character of this halogen displayed an important role for the effect observed by intraperitoneal route.
Administrated orally at 50 mg/kg, the compounds 3b and 3c reduced in 38.9% and 35.6%, respectively, the number of abdominal constrictions induced by acetic acid, suggesting that they can not be well absorbed by the gastrointestinal tract (Figure 2).
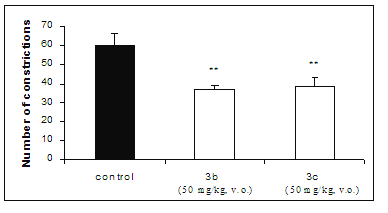
Figure 2: Effect on acetic acid-induced pain in mice test of compounds 3b and 3c, administrated orally at 50 mg/kg by. Each group represents the mean of six experiments. ** p < 0.01, compared with corresponding control value.
In addition, the potency of the compounds 3b and 3c in the writhing test (Table 3) exhibited a profile dose-dependent, presenting calculated ID50 values (in a 95% of confidence limit) of 40.8 (30.5 - 54.4) μmol/Kg and 30.8 (24.0 - 39.5) mmol/Kg, indicating that these compounds were ca of three to four times more active than acetylsalicylic acid and acetaminophen in the writhing assay.
Table 3: Comparison of analgesic effect between compound 3b and 3c and acetylsalicylic acid and acetaminophen, used as reference drugs.

The selected compounds 3b and 3c were also analyzed in the formalin-induced pain test, a reported behaviour model characterized by neurogenic (first phase) and inflammatory (second phase) phases (16) (Table 3). The results revealed that both, the δ-valerolactones studied and acetyl salicylic acid were inactive in preventing the first phase of the formalin-induced (neurogenic pain). However, 3b and 3c inhibited the inflammatory pain, 14.9% and 29.73% respectively, although both were less active that the reference drug in this second phase (Table 3). In addition, this experimental model permitted to evidenciate that the studied compounds do not reduce the paw oedema induced by this algic agent (results not shown).
The capsaicin test in mice has been employed to access the antinociceptive effect of tachykinin neurokinin-1 receptor antagonist, glutamate receptor antagonist, nitric oxide (NO) synthase inhibitor, and morphine (17). The δ-valerolactones 3b and 3c were tested intraperitoneally in mice, at 10 mg/Kg. The results of this test indicated that these compounds are ineffective on neurogenic pain, according the before observed, in the first phase of the formalin test (results not shown).
In the model of thermal sensibility, the compounds 3b and 3c, administrated i.p., compared with control group, delayed the reaction time on the hot plate. Thus, the mean reaction time ± S.E.M. was 8.9 ± 2.0 s in the control group, and this increased significantly to 19 ± 2.0 and 17 ± 1.0 s in those treated with 3b and 3c, respectively. The δ-valerolactones studied demonstrated to be effective in the hot-plate assay of nociception, suggesting to be related to the activation of opioid receptors, although they were less active than the morphine in the same assay (Figure 3). However, further studies are required to elucidate which mechanism of action is involved.
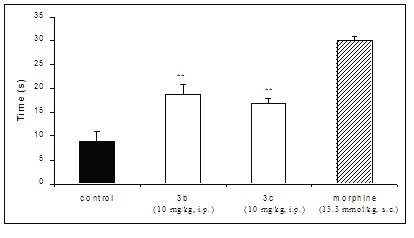
Figure 3: Effect of the compounds 3b and 3c (10 mg/Kg, i.p.) and morphine (13.3 μmol/Kg, s.c.) against hot plate-induced antinociception in mice. Each group represents the mean of six experiments. ** p < 0.01, compared with corresponding control value.
In summary, our results indicate that the studied δ-valerolactones present an interesting profile of analgesic action. These compounds were more potent than some clinically used drugs against writhing test. The elucidation of the mechanism of action needs additional studies, because the analgesic action of the studied compounds can be related to a peripheric action and/or to a neurogenic pain. Further studies should been necessary to elucidate the molecular events and pathway involved in pain process.
Experimental
CHEMICAL PROCEDURES
All IR spectra were recorded on a Shimadzu DR 8001 FTIR Spectrophotometer in transmittance mode, with 4 cm -1 resolution and with 40 accumulations (λ max in cm-1 ). The KBr pellets were prepared on a quartz mortar with about 1% of sample. The 1H-NMR and 13 NMR spectra were recorded using a Varian XL-200 at 200 MHz and 50 MHz, respectively. Chemical shifts are reported as δ values (ppm) relative to TMS (0.0 ppm). The correlation study HETCOR 1H-13 C and APT has also been performed. TLC was performed on the plates Kieselgel 60 F254 of Merck, using ethyl acetate and cyclohexane (1:1) as mobile phase. The melting points were determined using a Koefler apparatus and are uncorrected. Ethyl acetoacetate and other reagents were obtained from commercial sources. All solvents were distilled prior use.
PREPARATION OF COMPOUNDS 3a-e GENERAL PROCEDURE
Dried THF was bobbled with argon and cooled until 0 oC. Diisopropylamine (0.5 mL, 4.2 mmol) and n-buthyllitium were added under stirring and the mixture was left at 0°C for 45 minutes. The ethyl acetoacetate (0.2 mL, 1.5 mmol) was added drop wise for approximately 20 minutes. The dianion formation was observed by colour change of the reactional mixture. After that, the corresponding aldehyde (0.75 mmol) was added drop wise and the reaction stirred for 20 minutes, than cold water was added (25 mL) and the mixture was left to stand at room temperature for 3 hours. The crude mixture was extracted with Et2O (3 x 6 mL) and the aqueous layer acidified with hydrochloric acid (pH = 1) under ice bath. The solid thus obtained was filtered off, washed with water and Et2O to give 3a-3e; their yields and some physical constants are shown in Table 1.
4-oxo-6-( α-trans -styryl)-5,6-dihydro-2-pyrone (3a): FT-IR, KBr pellets: 1699 cm -1 , 1208 cm -1 ; 1 H NMR (DMSO-d6, ppm, 300 MHz): 2.57 (m, 2H, C-5H 2 ); 5.04 (m, 2H, C-3H, C-6H); 6.42 (dd, 1H, C-7H, 2 J 7a-7b = 16.0 Hz, 3 J 7a-6 = 6.2 Hz); 6.73 (d, 1H, C-8H, 2 J 7a-7b = 16.0 Hz); 7.25-7.51 (m, 5H, aromatic-H); 13 C NMR (DMSO-d6, ppm 75 MHz): 33 (C-5), 75 (C-6), 91 (C-3), 127, 128 and 129 (C-2', C-3', C-4', C-5' and C-6'), 127 (C-7), 132 (C-8), 136 (C-1'), 167 (C-2), 173 (C-4).
4-oxo-6-(phenyl)-5,6-dihydro-2-pyrone (3b): FT-IR, KBr pellets (cm -1 ): 1600, 1581, 1287; 1H NMR (DMSO-d6, ppm, 300 MHz): 2.7 (m, 2H, C-5H2); 5.11 (s, 1H, C-3H); 5.45 (m, 1H, C-6H); 7.38-7.44 (m, 5H, aromatic-H); 13 C NMR (DMSO-d6, ppm 75 MHz): 34 (C-5), 76 (C-6), 91 (C-3), 126, 128.3 and 128.5 (C-2', C-3', C-4', C-5' and C-6'), 139 (C-1'), 167 (C-2), 173 (C-4).
4-oxo-6-(4-fluorobenzene)-5,6-dihydro-2-pyrone (3c): FT-IR, KBr pellets (cm-1 ): 1600, 1581, 1208; 1H NMR (DMSO-d6, ppm, 300 MHz): 2.67 (m, 2H, C-5H2); 5.10 (s, 1H, C-3H); 5.7 (m, 1H, C-6H); 7.2-7.4 (m, 2H, C-2'H, C-6'H); 7.5-7.6 (m, 2H, C-3'H, C-5'H); 13 C NMR (DMSO-d6, ppm 75 MHz): 34 (C-5), 76 (C-6), 91 (C-3), 115.44 and 115.15 (C-2' and C-6'), 128.67 and 128.78 (C-3' and C-5'), 135 (C-1'), 162 (d, C-4, J CF = 244 Hz), 167 (C-2), 173 (C-4).
4-oxo-6-(isopropyl)-5,6-dihydro-2-pyrone (3d): FT-IR, KBr pellets (cm-1 ): 1732, 1626, 1266; 1H NMR (DMSO-d6, ppm, 300 MHz): 0.93 (m, 6H, CH 3 ); 1.86 (m, 1H, C-7H); 2.35 (m, 2H, C-5H 2 ); 4.07 (m, 1H, C-6H); 4.94 (s, 1H, C-3H); 13 C NMR (CDCl3, ppm 75 MHz): 20 (2C, CH 3 ), 32 (C-7), 40 (C-5), 45 (C-3), 80 (C-6), 168 (C-2), 200 (C-4).
4-oxo-6-(isobutyl)-5,6-dihydro-2-pyrone (3e): FT-IR, KBr pellets (cm-1 ): 1700, 1641, 1217; 1H NMR (DMSO-d6, ppm, 300 MHz): 0.85 (m, 6H, CH 3 ); 1.3 (m, 2H, C-7H 2 ); 1.7 (m, 1H, C-8H); 2.35 (m, 2H, C-5H 2 ); 3.9 (m, 1H, C-6H); 4.5 (s, 1H, C-3H); 13 C NMR (CDCl 3 , ppm 75 MHz): 17 (1C, CH 3 ), 18 (1C, CH 3 ), 30 (C-7), 32 (C-8), 41 (C-5), 47 (C-3), 80 (C-6), 167 (C-2), 201 (C-4).
PHARMACOLOGICAL ASSAYS
ANIMALS
Male Swiss mice (25 - 35 g), housed at 22 ± 2 oC under a 12 h light/12 h dark cycle and with access to food and water ad libitum, were used. Experiments were performed during the light phase of the cycle. Animals were acclimatized to the laboratory for at least 2 h before testing and were used once throughout the experiments. All experiments reported in this study were carried out in accordance with current guidelines for the care of laboratory animals and ethical guidelines for investigation of experimental pain in conscious animals (18).
DRUGS
The following substances were used: acetic acid, formalin, capsaicin (Calbiochen, San Diego, CA, USA), morphine hydrochloride (Merck, Darmstadt, Germany). The δ-valerolactones as well as reference drugs were dissolved in Tween 80 (E. Merck), plus 0,9% of NaCl solution, with the exception of capsaicin which was dissolved in ethanol. The final concentration of Tween 80 and ethanol did not exceed 5% and did not cause any effect `per se'.
WRITHING TEST
The abdominal constriction was induced in mice by intraperitoneal injection of acetic acid (0.6%), as described by Collier et al. (1968) with minor modifications (19). Animals were pre-treated intraperitoneally (10 mg kg -1 , 30 min before) and orally (50 mg kg -1 , 60 min before) with the δ-valerolactones. Control animals received a similar volume of saline solution (10 ml kg -1 ). The number of abdominal constrictions (full extension of both hind paws) was cumulatively counted over a period of 20 min. Antinociceptive activity was expressed as the reduction of the number of abdominal constrictions between control animals and mice pre-treated with δ-valerolactones.
FORMALIN TEST
The observation chamber was a glass cylinder of 20 cm diameter with a mirror at a 45. angle to allow clear observation of the paws of the animals. The mice were treated with 0.9% saline solution (i.p.), or δ-valerolactones (10 mg kg -1 , i.p.) 30 min before formalin injection. Each animal was placed in the chamber for 5 min before treatment in order to allow acclimatization to the new environment. The formalin test was carried out as described by Hunskaar and Hole (1987) with minor modifications (16). Twenty micro litres of a 2.5% formalin solution (0.92% formaldehyde) in 0,9% saline solution were injected intraplantarly in the right hind paw. The animal was then returned to the chamber and the amount of time that it spent licking the injected paw was considered as indicative of pain. Two distinct phases of intensive licking activity were identified: an early acute phase and a late or tonic phase (0-5 and 15-30 min after formalin injection, respectively). At the end of the experiments the animals were sacrificed by cervical dislocation and the paws cut at the tibio-tarsic joint and weighed on an analytical balance to investigate the interference of the marrubiin on formalin-induced inflammatory oedema.
HOT-PLATE TEST
The hot-plate test was used to measure response latencies. The mice were treated with saline solution, morphine (10 mg kg -1 , s.c.) or the δ-valerolactones (10 mg kg -1 , i.p.) placed individually on a hot plate maintained at 56±1 oC and the time between placement of the animal on the hot plate and the occurrence of either the licking of the hind paws, shaking or jumping off from the surface was recorded as response latency. Mice with baseline latencies of more than 20 s were eliminated from the study and the cut-off time for the hot-plate latencies was set at 30 s. The animals were treated 30 min before the assay.
STATISTICAL ANALYSIS
The results are presented as mean ± S.E.M., except the ID values (i.e. the dose of marrubiinic acid reducing the 50 nociceptive response by 50% relative to the control value), which are reported as geometric means accompanied by their respective 95% confidence limits. The ID value was 50 determined by linear regression from individual experiments using linear regression GraphPad software (GraphPad software, San Diego, CA). The statistical significance of differences between groups was detected by ANOVA followed by Dunnett's multiple comparison tests. P-values less than 0.05 (P,0.05) were considered as indicative of significance.
Acknowledgements
The authors are grateful to CNPq for P. Amaral and CAPES/COFECUB for E. M. Santos by financial support (scholarship). We wish to thank Prof. Jairton Dupont and Joyce C. Espinola for NMR analyzes (Instituto de Química/UFRGS).
References
Sliskovic D.R., Roth B.D., Wilson M.W., Hoefle M.L. and Newton R.S. Inhibitors of cholesterol biosynthesis. 2. 1,3,5-Trisubstituted [2-(tetrahydro-4-hydroxy-2-oxopyran-6-yl)ethyl]pyrazoles. J. Med. Chem., 33: 31-38, 1990.
Skaltsa, H., Lazari, D., Panagoulcas, C., Sokovic, Garcia, E. B., and Sorovic, M. Sesquiterpene lactones from Centaurea thessala and Centaurea attica with Antifungal activity. Phytochem., 55: 903-8, 2000.
Tait, B.D., Hagen, S., Domagala, J., Ellsworth, E., Gadja, C., Hamilton, H., Vara Prasad, J.V., Graham, N., Hupe, D., Nouhan, C., Tummino, P., Humblet, C., Lunney, E., Pavlovsky, A., Rubin, J., Gracheck, S., Baldwin, E., Bhat, T., Erickson, J., Gulnik, S. and Liu, B. 4-Hydroxy-5,6-dihydropyrones Potent Non-Peptide Inhibitors of HIV Protease. J. Med. Chem., 40: 3781-3792, 1997.
Malsch, U. and Kieser, M. Efficacy of kava-kava in the treatment of non-psychotic anxiety, following pre-treatment with benzodiazepines. Psychopharmacol., 157: 277-283, 2001.
Fowler E.M.F. and Henbest, H.B. Researches on Acetylenic Compounds. Part XXV. Synthesis of (±)–Kawain. Chem. Soc., 3642-3645, 1950.
Cho, J.Y., Bayk, K.U., Jung J.H. and Park, M.H. In vitro anti-inflammatory effects of cynaropicrin, a sesquiterpene lactone from Saussurea lappa. Eur. J. Pharmacol., 399-407, 2000.
Le Hetet, C., David, M., Carreaux, F., Carboni, B. Sauleau, A. Synthesis of functionalised g- and d-lactones via polymer-bound epoxides. Tetrahedron Lett., 38: 5153-5156, 1997.
Gouault, N, Cupif, J.F., Sauleau, A. and David, M. g-Methyl-substituted-g-butyrolactones: solid-phase synthesis employing a cyclisation-cleavage strategy. Tetrahedron Lett., 41: 7293-7297, 2000.
Harris, J.M. and O'Doherty, G.A. An olefination approach to the enantioselective syntheses of several styryllactones. Tetrahedron, 57:5161-5171, 2001.
Kiegiel J., Jówik J., Woniak K. and Jurczak, J. Synthesis and asymmetric hydrogenation of 3,5-dioxoheptanedioates. Preparation of enantiomerically pure substituted d-valerolactones, Tetrahedron Lett., 41: 4959-4963, 2000.
Reffstrup, N. and Boll, P.M. The Preparation of 4-Hydroxy-5,6-dihydro-2-pyrones and Their Conversion to Kawa-lactones as well as to Other Precursors of Naturally Occurring 2-Pyrones. Acta. Chem. Scan. B-Org Chem. Biochem., 30: 613-617, 1976.
Dharmaratne, H.R.W., Nanayakkara, N.P.D. and Khan, I.A. Kavalactones from Pipernmethysticum, and their 13C NMR spectroscopic analyses. Phytochem., 59: 429-433. 2002.
Calixto J.B., Santos A.R., Cechinel Filho, V. and Yunes R.A. A review of the plants of the genus Phylanthus: their chemistry, pharmacology, and therapeutic potential. Med. Res. Rev., 18, 225-258, 1998.
De Jesus, R.A.P., Cechinel Filho, V. and Oliveira, A.E. Analysis of the antinociceptive properties of marrubiin isolated from Marrubium vulgare. Phytomed., 2:111-5, 1999.
Ochi, T., Motoyama, Y. and Goto, T. The analgesic effect profile of FR122047, a selective cyclooxygenase-1 inhibitor, in chemical nociceptive models. Eur. J. Pharmacol., 1-2:49-54, 2000.
Hunskaar, S. and Hole, K. The formalin test in mice: dissociation between inflammatory and non-inflammatory pain. Pain, 30:103-114, 1987.
Sakurada T., Matsumura, T., Moriyama, T., Sakurada, C., Ueno, S. and Sakurada, S. Differential effects of intraplantar capsazepine and ruthenium red on capsaicin-induced desensitization in mice. Pharmacol. Biochem. Behav., 75:115-21, 2003.
Zimmermann, M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain, 16:109-110, 1983.
Collier, H.O., Dinneen, L.C., Johnson, C.A. and Schneider, C. The abdominal constriction response and its suppression by analgesic drugs in the mouse. Br. J. Pharmacol., 32:295-310, 1968.
Corresponding Author: V. L. Eifler-Lima. Faculty of Pharmacy, Federal University of Rio Grande do Sul - UFRGS, Av. Ipiranga 2752, Porto Alegre/RS, Brazil, ZIP code: 90610-000. eifler@farmacia.ufrgs.br www.ufrgs.br/farmacia/laboratorios/lsf/index.htm
Published by the Canadian Society for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences.
http://www.cspscanada.org