Effect of efavirenz on intestinal p-glycoprotein and hepatic p450 function in rats.
Nathalie Berruet, Stephanie Sentenac, Daniel Auchere, François Gimenez, Robert Farinotti, Christine Fernandez
Department
of Clinical Pharmacy, Faculty of Pharmacy, EA 2706,
Received February 5, 2005, Revised April 6, 2005, Accepted May 9, 2005, Published August 4, 2005
ABSTRACT. Purpose: P-glycoprotein (P-gp) and cytochrome P450 (P450) affect drug disposition. Efavirenz (EFV) is an anti-HIV drug used in combination. Since most anti-HIV medications are substrate and modulators of P-gp and/or P450, we investigated the effects of EFV on intestinal P-gp and hepatic P450 function to predict drug interactions. Methods: (i) The effect of EFV on rat intestinal P-gp function was studied on everted gut sacs and in situ intestinal perfusion. EFV was orally administered (150 mg/kg) for 6 days. Then, rhodamine 123 was used as a P-gp substrate and verapamil as an inhibitor. P-gp function was evaluated by the difference between rhodamine 123 transport with and without verapamil. (ii) The effect of EFV on rat hepatic P450 metabolism was investigated using hepatic microsomes, prepared from rats pretreated or not with EFV. RESULTS : In everted gut sacs, P-gp function was not modified and in the in situ intestinal perfusion, rhodamine 123 clearance was not affected by EFV. Concentrations of the metabolites, 1-OH midazolam and 4-OH midazolam were higher in EFV pretreated rats than those in the control group. Conclusion: EFV should not modify intestinal absorption of co-administered substrates of P-gp, but could decrease plasma concentrations of co-administered drugs metabolized by P450.
INTRODUCTION
Efavirenz (EFV) is a non nucleoside reverse transcriptase inhibitor (NNRTI) used in the treatment of HIV-1 infection with favorable safety and pharmacokinetics. Its therapeutic use can be limited by the rapid emergence of resistant viruses as the result of single nucleotide changes in monotherapy. Hence, EFV is used in combination with nucleoside analogs or protease inhibitors. Its good bioavailability and its long terminal half-life in humans permit once-daily dosing. EFV is extensively metabolized by P450 isoenzymes, mainly 3A4 and 2D6 (1). Following oral administration, less than 1% of the dose is excreted unchanged in urine. EFV is an inhibitor of P450 isoenzymes (2C9, 2C19, 3A4 and 1A2) and can also induce 3A4 and 2D6 isoenzymes (2). In humans, EFV increases AUC and Cmax of ritonavir (17% and 24%, respectively). On the opposite, it decreases saquinavir AUC and Cmax (60% and 50%, respectively) (3). It was also reported that, in humans, EFV could induce hepatic, but not intestinal CYP3A4 (1).
P-glycoprotein (P-gp) is a member of the superfamily of ATP-binding cassette (ABC) transport proteins. It plays an important role in transport of many different substrates, including antiretroviral drugs (4-6). P-gp is constitutively expressed and abundant in the apical membrane of many epithelial and endothelial barriers such as the intestine, the kidney, the blood-brain, blood-testis and materno-fœtal barriers (7-8). P-gp is encoded by MDR1 and MDR2 genes in humans and mdr1a, mdr1b and mdr2 genes in rodents. Variation of expression of these genes can lead to differences in drug exposure and, therefore, modify virological and immunological responses to antiretroviral treatment. There is a significant overlap in substrate specificity and modulation between P-gp and P450 (9).
We investigated the effects of EFV on intestinal P-gp and hepatic P450 functionalities in order to predict drug interactions with EFV. The effect of EFV on intestinal P-gp function was studied on two models (everted gut sac, in situ intestinal perfusion) using rhodamine 123 as P-gp substrate and repeated oral doses of EFV as P-gp modulator. The effect of EFV on P450 hepatic metabolism was investigated on hepatic microsomes, extracted from rats pre-treated with EFV or not and using midazolam as substrate (metabolized in rats, mainly into 4-OH midazolam, by P450 3A1 and 3A2)(10). Verapamil was used as a P-gp inhibitor.
MATERIALS AND METHODS
Materials. Efavirenz (SUSTIVAâ), midazolam (HYPNOVELâ), 1-OH and 4-OH midazolam, pentobarbital (PENTOBARBITAL SODIQUEâ) were purchased from Dupont Pharma (New York, USA), Roche (Neuilly/Seine, France), Ultrafine Chemicals (Manchester, UK), and Sanofi-Synthelabo (Libourne, France); respectively. Verapamil, prazepam, rhodamine 123 and carboxymethylcellulose were purchased from Sigma-Aldrich (Saint Quentin Fallavier, France). All other reagents were of analytical grade from local suppliers.
Animals. Male adult Sprague Dawley rats (weight : 280 to 305 g) were purchased
from Iffa-Credo (L’Abresle, France). In vivo experimentations were performed
in accordance with the guidelines issued by the
In P-glycoprotein and P450 induction assays, EFV was administered by oral route (150 mg/kg) for 6 days before experiments. A 24 hour wash out period was respected before experiments in oder to avoid direct interactions between EFV and the other substrates (rhodamine 123, midazolam).
Preparation of everted gut sacs for in vitro study of Rhodamine 123 transport. Rats were treated with efavirenz (150 mg/kg) (n=6) or with the vehicle alone (0.2% carboxymethylcellulose) (n=6) once daily, by oral route, for 6 days. They were fasted overnight (with free access to water). After laparotomy, the small intestine was flushed with cold 0.9% saline and removed. A 10 cm segment was collected at the jejunum level (after the ligament of Treitz), ligated at one extremity, everted and filled with a 270 µM solution of Rhodamine 123 with or without a P-gp inhibitor. Verapamil was chosen as inhibitor, as it was previously described in this model (11) (however, verapamil was also described by other authors as an MRP1 and MRP4 inhibitor (12-13)). The other extremity was ligated and the sac was incubated at 37°C, in a Krebs-Ringer-sodium bicarbonate pH 7.4 buffer. Verapamil was added to the serosal and the mucosal sides, at the same concentration (250µM).
In order to study the efflux function, rhodamine 123 transport from the serosal to the mucosal side was measured by collecting 1 ml of the buffer every 20 minutes from 0 to 100 minutes (14). This model was validated on animals pre-treated with dexamethasone (40 mg/kg during 3 days) by oral route: 3 rats received dexamethasone and 3 rats received the vehicle alone (20% tween 20 solution). Rhodamine was quantified by direct spectrofluorimetry (excitation wavelength: 500 nm, emission wavelength: 525 nm). The transport of Rhodamine 123 from serosal to mucosal surfaces across the sac was claculated using Equation 1:
Amount of rhodamine in the buffer (mucosal
side) |
1 |
||
-------------------------------------------------- |
x 100 x |
--------------------- |
(%/min) ( eq. 1) |
Initial amount of Rhodamine in the sac (serosal
side) |
incubation time (min) |
In situ rat intestinal perfusion. Intestinal perfusion was performed as previously reported (10). Rats were treated with EFV (150 mg/kg) (n=6) or the vehicle alone (0.2% carboxymethylcellulose) (n=6) once daily, by oral route, for 6 days. Anesthesia was induced with an intraperitoneal injection of 1.5 g/kg urethane. A midline longitudinal incision was made. Inlet and outlet canulas were inserted in 20 cm of the jejunum. The inlet tubing was connected to an infusion pump. Intestinal effluent samples were collected into heparinized vials at 20, 40 and 60 min. Blood samples were withdrawn from the carotid 10, 30 and 50 minutes after the start of the intestinal perfusion.
Intestinal perfusion was done for up to 30 minutes before equilibrium (0.01M pH7.4 phosphate buffer,0.3 ml/min). Meanwhile, NaCl 0.9% was perfused in the jugular vein (2 ml/min). Thirty minutes after the beginning of the perfusion, a bolus of rhodamine 123 (4.36 ml/kg of a 100 µM solution), followed by an infusion (100 µM at 2 ml/h) were done. After 110 min of jugular perfusion, the intestinal segment was collected and its surface was measured.
At the end of the experiments, rats were euthanized by a intraperitoneal injection of pentobarbital.
Intestinal rhodamine clearance (Cl Rh) was calculated as follows:
CI F
Cl Rh = ------- x ------ 1000 (µl/min/cm²) ( eq.2)
Cp S
(CI : concentration of rhodamine 123 in the perfusate (µg/ml), Cp : concentration of rhodamine 123 in plasma (µg/ml), F : perfusion flow rate (ml/min) and S : surface of the intestinal segment (cm²)).
Rhodamine 123 was quantified by liquid chromatography (spectrofluorimetric detection : lexcitation: 500 nm, lemission: 525 nm) (15) .
Cytochrome P450 activity. Rats were treated with EFV (150 mg/kg) (n=6) or the vehicle alone (0.2% carboxymethylcellulose) (n=6) once daily, by oral route, during 6 days. One gram of frozen liver was homogenized in 2 ml of pH 8 sodium phosphate buffer (50 mM) containing saccharose (0.25 M), EDTA (10 mM) and dithiotreitol (0.1 mM). The preparation was centrifuged 10 min at 1500 g and 30 min at 100 000 g. The pellet was discarded and the supernatant was centrifuged at 100 000 g for 60 min. The pellet was resuspended in 100 mM pH 8 potassium phosphate buffer containing EDTA (1 mM) and dithiotreitol (1mM). The suspension was stored at –80°C until use. Incubation media were composed of pH7.4 phosphate buffer (0.1M), NADPH (1 mM), and midazolam (0, 1, 2, 5, 10, 25, and 50 µM). Protein concentrations were determined using Bradford’s method (16) . Hepatic microsome final concentration was set at 0.4 mg/ml.
Midazolam and microsomes were preincubated at 37°C for 5 minutes. The reaction was initiated by the addition of NADPH,H+ and terminated by the addition of 200 µl of ice cold methanol containing the internal standard (prazepam). The mixture was homogenized and centrifuged at 3000 g for 10 minutes. One hundred µl of the supernatant were injected onto the chromatographic system for analysis. Midazolam, 1-OH midazolam and 4-OH midazolam were quantified as described in the “midazolam analysis” section.
Data for the individual metabolites were fitted and
Km and Vmax were calculated for each individual dataset using the Sigmaplot-Enzyme
Kinetics software (SPSS,
Clint = Vmax x Km-1 x microsomal protein content (g liver-1) x g liver kg-1
Midazolam analysis. Midazolam concentrations were determined by liquid chromatography (UV detection l=230 nm). One hundred and forty µL of 0.1N sodium hydroxyde, 2 ml of ethyl acetate, 40 µl of prazepam (internal standard, 1µg/ml in water) and 100 µl of sample were shaken for 10 minutes and then centrifuged at 3000 rpm (10 minutes).The organic phase was evaporated. The residue was dissolved in 100 µl of methanol and 50 µl were injected into a Symmetry C18 column (150 x 3.9 mm, id, Waters, Milford, MA, USA) at a flow rate of mobile phase (methanol / 0.1M pH7.4 sodium phosphate buffer / water / acetonitrile, 65:10:24:1, v/v/v/v) of 1 ml/min.
Statistics. Clearance of rhodamine 123 values, Km, Vmax and Clint results were compared using a non parametric Mann –Whitney test (p = 0.05).
RESULTS
The dose of EFV (150 mg/kg) was chosen in order to obtain plasmatic concentrations still detectable 24 hours after administration (results not shown). No difference was found in ALAT/ASAT, creatinine and total protein values between EFV pre-treated and non pre-treated rats.
Transport of rhodamine 123 in everted gut sacs. The clearance of rhodamine 123, expressed as the percentage of rhodamine 123 transported from the serosal to the mucosal compartment, is presented in figures 1 and 2 for rats pre-treated with dexamethasone and EFV, respectively.
In the dexamethasone experiments, a statistical difference (p<0.05) was found in the percentage of Rhodamine 123 transported between pre-treated and non pre-treated animals (figure 1A) showing that dexamethasone induces P-gp function. With verapamil, the percentage of rhodamine 123 transported was decreased, not significantly, between dexamethasone pre-treated and non pre-treated animals (figure 1B), showing that verapamil can not completely inhibit P-gp function if it has been previously induced.
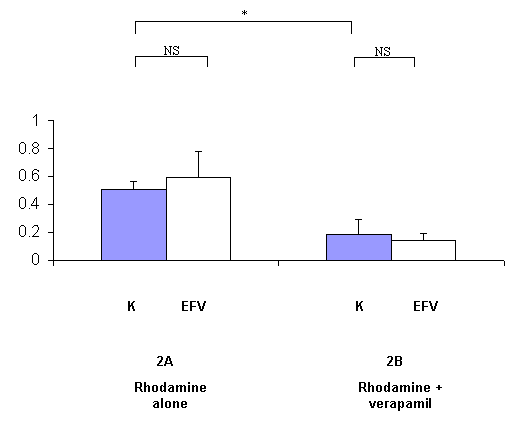
In the EFV experiments, no statistical difference was evidenced between pre-treated and non-pre-treated animals (figure 2A), showing that EFV does not induce P-gp function. The transport of rhodamine 123 was decreased by verapamil by a 2.5-fold factor between EFV non-pre-treated and EFV pre-treated animals.
In situ rat intestinal perfusion. We did not show any effect of EFV pre-treatment on rhodamine 123 clearance: in intestinal perfusion assays, rhodamine 123 clearance was found to be 15.01 (+/- 8.99) and 14.77 (+/- 6.89) µl/min/cm² in non pre-treated (n=6) and pre-treated rats (n=6), respectively.
Midazolam metabolism. Km, Vmax and Clint values for the formation of 4-OH midazolam and 1-OH midazolam are presented in Table 1. These results show that EFV induces midazolam metabolism.
Figure 1: Effect of dexamethasone (Dex) on the clearance of rhodamine 123 (Rh), expressed as the percentage of rhodamine 123 transported across rat everted gut sacs without (1A, n=3) or with a P-gp blocker (verapamil) (1B, n=3) per minute. Grey columns represent rhodamine 123 transport in control rats non pre-treated with dexamethasone (K) and white columns represent rhodamine 123 transport in rats pre-treated with dexamethasone (40 mg/kg during 3 days) (Dex) (m +/- s.d.). *: p<0.05 ; NS : non significant.
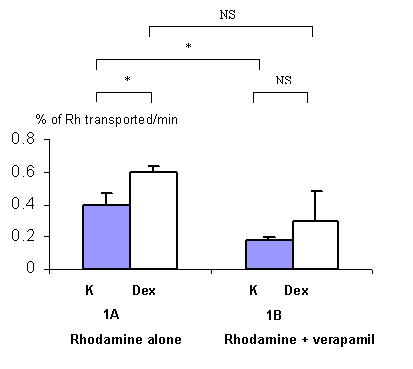 |
Figure 2: Effect of efavirenz (EFV) on the clearance of rhodamine 123 (Rh), expressed as the percentage of rhodamine 123 transported across rat everted gut sacs without (2A, n=6) or with a P-gp blocker (verapamil) (2B, n=7) per minute. Grey columns represent rhodamine 123 transport in control rats non pre-treated with EFV (K) and white columns represent rhodamine 123 transport in rats pre-treated with EFV (150 mg/kg during 6 days) (EFV) (m +/- s.d.). *: p<0.05 ; NS : non significant
DISCUSSION
As the duration of pre-treatment is an important factor for modulation of P-gp and P450, we have chosen a 6 day pre-treatment by EFV in order to ensure hepatic and intestinal cellular turnover. Our experiments showed that oral repeated doses of EFV do not modify intestinal P-gp function in rats. On the contrary, EFV induces hepatic P450 dependent metabolism.
Modulation of intestinal P-glycoprotein by EFV. Regarding modulation of P-gp by EFV, controversial reports exist in the literature. These data result from different experimental models. Differences between EFV concentrations tested or cellular models used may explain some of the discrepancies in results. Our results confirmed those of Mouly et al who did not evidence any induction of intestinal P-gp expression by EFV working on human intestinal biopsies (1). On the contrary, on P-gp-expressing LS180v cells, Stormer et al showed that EFV was not a P-gp substrate but that, after a 6-day treatment, EFV was able to slightly induce P-gp functionality (17).
On peripheral blood mononuclear cells, Chandler et al showed that EFV could induce P-gp expression. However, the authors hypothetised that P-gp upregulation could be due to cell stress induced by EFV cell toxicity (18).The ex-vivo model (everted gut sacs), previously described and used by others (19), was validated by a 3-day pre-treatment with dexamethasone which provoked an induction of P-gp. Conversely, after a 6-day treatment with EFV, no induction was observed. Because of the large inter individual variability, evidencing an effect of a mild inducer or a mild inhibitor would require a larger number of animals (20) than the one used in our experiments. For this reason, we decided to confirm our results using the intestinal perfusion model which respects blood perfusion.
As Veau et al (15) have shown that renal failure can reduce intestinal P-gp functionality, we have verified that EFV did not induce any renal alteration as previously reported by Mutlib et al (21), by checking creatininemia. This model confirmed that EFV does not modify intestinal clearance of rhodamine 123, a non metabolized major substrate of P-gp.
Modulation of hepatic metabolism by cytochromes P450. Midazolam was chosen in our experiments because it is metabolized by P450 3A1 and 3A2 enzymes in rats but was described as a non-substrate of P-gp (22-24). We observed an induction of hepatic P450 3A1 and 3A2 by EFV in vitro.
Mouly et al demonstrated that EFV could induce P450 3A4 in the liver but not in the intestine (1). Duration of exposure of EFV to the enterocyte during oral treatment was insufficient to cause an increase in gene transcription and, therefore an induction of intestinal P450 and P-gp (1). For this reason, we have studied hepatic P450 enzymes.
In vitro, on isolated hepatic microsomes, 4-OH midazolam concentrations were higher in rats pre-treated with EFV than in the control group. In vitro kinetic data (Vmax, Km and Clint) for non-pretreated rats were similar to that previously reported by other authors (10) under similar experimental conditions. Our assays evidenced an induction of P450 enzymes and specifically of 3A1 and 3A2 isoforms. This effect was also observed in humans by an increased formation of 1-OH midazolam preferentially produced by P450 3A4 (not present in rodents) and 3A5 (25). However, the effects of enzyme or efflux protein modulators can be tissue-dependent, as the expression of these proteins is also tissue-specific.
TABLE 1: In vitro data of midazolam metabolism (expressed as conversion of midazolam to 4-OH and 1-OH midazolam) by liver microsomes prepared from control rats (n=6) or pretreated rats (EFV 150 mg/kg during 6 days).
Control group |
EFV pre-treated group |
|
| 4-OH midazolam Vmax (µmol/mg of proteins/min) Km (µM) Clint (ml/min/mg of proteins) |
0.72 +/- 0.04 4.88 +/- 0.89 0.15 +/- 0.03 |
2.82 +/- 0.19 * 8.40 +/- 1.03 * 0.34 +/- 0.01 * |
| 1-OH midazolam Vmax (µmol/mg of proteins/min) Km (µM) Clint (ml/min/mg of proteins) |
0.28 +/- 0.02 5.22 +/- 0.96 0.06 +/- 0.01 |
0.90 +/- 0.07 * 5.43 +/- 0.73 0.16 +/- 0.02 * |
P-glycoprotein and cytochromes P450 modulation. There is an important overlap in substrate specificy between both systems and several substrates/modulators of P-gp are also substrates/modulators of P450 enzymes (26). For instance, saquinavir, an HIV protease inhibitor used in the treatment of HIV infection, is metabolized by P450 3A and transported by P-gp (27-29). However, there is no correlation between the potency of P-gp inhibition and CYP3A inhibition: ratios of IC50 for CYP3A to IC50 for P-gp may vary from 1.1 to 125 for inhibitors such as cyclosporine, quinidine or PSC833 (30). In some cases, a low expression of P-gp could be associated with a higher rate of metabolism. For instance, Fellay et al have shown that decresed P-gp expression in peripheral mononuclear cells was associated with low plasma concentrations of EFV, suggesting that other transporters could be over-expressed and/or that intestinal CYP450 could compensate the low P-gp expression (31). On the contrary, Winzer et al have found opposite results on a smaller group of patients : they did not evidence any relation between MDR1 3435 genotype and EFV plasma concentrations (32).
CONCLUSION
EFV was already known as a CYP3A4 inductor in humans. We have confirmed that EFV was also an inducer of hepatic CYP3A in rats and have shown, using an ex-vivo and an in vitro model, that EFV is not a modulator of intestinal P-gp. When combined to other drugs used to treat HIV infection, EFV should not modify their intestinal absorption rate by P-gp modulation but drug interactions with P-gp substrates can be sustained by the metabolic step.
REFERENCES