J Pharm Pharmaceut Sci (www.cspscanada.org) 8(2):175-181, 2005
Enhancing dissolution, serum concentrations and hypoglycemic effect of glibenclamide using solvent deposition technique.
Siavoush Dastmalchi*1, Alireza Garjani2, Nasrin Maleki2, Golaleh Sheikhee3, Vida Baghchevan2, Parisa Jafari-Azad2, Hadi Valizadeh3, Mohammad Barzegar-Jalali3
1Department of Medicinal Chemistry, School of Pharmacy, Tabriz University of Medical Sciences, Tabriz, Iran
2Department of Pharmacology, School of Pharmacy, Tabriz University of Medical Sciences, Tabriz, Iran
3Department of Pharmaceutics, School of Pharmacy, Tabriz University of Medical Sciences, Tabriz, IranReceived 7 March 2005, Revised 14 March 2005, Accepted 23 March 2005, Publlished 2 August 2005
Abstract
PURPOSE: Glibenclamide is practically insoluble in water and its GI absorption is limited by its dissolution rate. Therefore, to enhance the drug dissolution, serum concentrations and its hypoglycemic effects, it was formulated as solid dispersions and evaluated the relevant in vitro and in vivo parameters. METHODS: The drug solid dispersions were prepared by solvent deposition technique using microcrystalline cellulose as the carrier in different ratios and their dissolution rates were compared to those of pure drug and its physical mixture with carrier. Drug serum concentrations and hypoglycemic effects in rabbits of pure drug, a physical mixture and the corresponding solid dispersion were investigated. In order to elucidate the observed in vitro and in vivo differences, IR spectroscopy and x-ray diffraction patterns of the formulations were studied. RESULTS: The solid dispersion with the drug to carrier ratio of 1:19 showed the highest dissolution rate with the dissolution efficiency (DE) of 44.42 in comparison to pure drug (DE = 3.82), physical mixture (DE = 4.91) and other solid dispersions (DE between 13.85-39.94) and also produced higher drug serum concentrations (more than 4 times at 6th hour post dose) as well as enhanced hypoglycemic effects relative to pure drug and its corresponding physical mixture. CONCLUSIONS: Solvent deposition technique was proved an effective tool of increasing dissolution probably due to enhanced wettability and reduced drug particle size, which in turn led to enhance drug serum concentrations and its hypoglycemic effects. Strong quantitative correlations were established between dissolution parameter and parameters related to serum concentrations as well as hypoglycemic effects.
Introduction
Glibenclamide is a second generation sulfonylurea used in the treatment of nonisulin dependent diabetes. Its hypoglycemic effect is mainly due to stimulation of insulin release from pancreatic beta cells and sensitization of the peripheral tissues to insulin (1).
Glibenclamide is practically insoluble in water (1) which leads to poor dissolution rate and subsequent decrease of its gastrointestinal (GI) absorption. Results of several investigations revealed that the absorption of glibenclamide was limited by its dissolution rate (2-9). The solid dispersion technique has been widely used to enhance dissolution rate of glibenclamide (3, 4, 7-9). In the present work, we have used solvent deposition (SD) technique to prepare glibenclamide solid dispersion in microcrystalline cellulose as the carrier at different ratios of drug to carrier. To the best of our knowledge, the solvent deposition technique and the carriers used in this work have not been applied to this drug. After performing the dissolution tests on pure drug (PD), physical mixture (PM) and SD formulations, x-ray diffraction pattern and infrared (IR) spectroscopic method have been employed to elucidate possible crystal changes in glibenclamide and drug-carrier interactions. Then the solid dispersion with the fastest dissolution rate was chosen for the further assessment of its serum concentrations as well as hypoglycemic effect in the rabbits.
Materials and Methods
Glibenclamide (Chinoin Pharma, Hungaria), microcrystalline cellulose (Avicel PH-102 and RC591, FMC, Brussels, Belgium), chloroform, potassium dihydrogen phosphate and disodium hydrogen phosphate (Merck, Darmstadt, Germany). Glucose assay kit (Darman Kave Res Lab, Isfahan, Iran).
Preparation of SD systems and physical mixture
Solvent deposition (SD) systems were prepared by dissolving glibenclamide in chloroform to produce a clear solution (10, 11), then the carrier was dispersed in the solution by stirring at 38±0.5°C and the solvent was removed by evaporation at 61±0.5°C while stirring. The resultant mass was dried at 40°C for 24 hours, pulverized and passed through a sieve with a mesh number of 120. Four different ratios of drug to carrier, namely 1:1, 1:5, 1:9 and 1:19 were used for SD formulations. A physical mixture containing one part drug and nineteen parts carrier was prepared using the bottle method.
The details of formulations are given in Table 1. From each batch four samples each equivalent to 20 mg of glibenclamide were taken and dissolved in methanol to reach appropriate dilution and then analyzed for the drug content by UV spectroscopy at 228 nm (UV-160 Shimadzo, Kyoto, Japan) using an appropriate Beer's plot. The measured contents of glibenclomide in the physical mixture and the SD preparations were 20.05±0.1 mg.
Table 1: Dissolution effeciency of different formulations.
Dissolution rate studies
The dissolution rate of glibenclomide in powder form, physical mixture and SD systems was studied using Levy's beaker and stirrer method. Briefly, a sample equivalent to 20 mg of glibenclomide was added to 1000 ml of dissolution medium (phosphate buffer pH 7.25) and the mixture was stirred at 80 rpm with a two-bladed stirrer, 7.5 cm in diameter positioned 4 cm from the bottom of the beaker, at 37±0.3°C. Five millilitre samples of dissolution medium were withdrawn and filtered at different time intervals and assayed at 228 nm using a calibration curve. The drug concentration in each sample was corrected considering the concentrations in the previous samples. Each dissolution test was performed in triplicates.
X-ray crystallography and IR spectroscopy
X-ray diffraction patterns of the samples were obtained using an automatic powder diffractometer (Simens-850, Munich, Germany) using Cu Ka radiation at a scan rate of 2° min-1 in terms of 2q angle. The KBr disk sample preparation technique was used to obtain the IR spectra of the formulations on an IR spectrophotometer (FTIR 3400 Shimadzo, Kyoto, Japan).
Glibenclamide serum concentration
Ten white male healthy albino rabbits, 2.0±0.5 kg, were selected for this study. The permission for animal studies was obtained from the ethics committee of Tabriz University of Medical Sciences. Twelve hours before the experiment, the animals were fasted but had free access to tap water. Each of the animals received a single dose of glibenclamide (equivalent to 2 mg/kg) as pure drug, physical mixture of drug and Avicel PH102 (1:19) and its SD (1:19) in an aqueous suspension form according to a three-treatment randomized crossover schedule. Thus, each animal received three preparations on three treatment days with a two-week washout period between two successive dosing. The suspensions were prepared by dispersing the pure glibenclamide powder or its SD and PM preparations in 10 ml distilled water and administered in the esophagus with a syringe equipped with a number 12 catheter. Blood samples were collected predose (0 hr) and 1, 2, 3, 4, 5 and 6 hr post dose from marginal vein through a catheter inserted in the vein. Since, in the ascending part of the drug serum concentration curve, the absorption rate is dominant compared to elimination rate and the dissolution rate is the major factor affecting the serum concentrations, attempts were not made to collect the blood samples beyond six hours. After clot retraction, the samples centrifuged at 3500 rpm for 15 min, and then the serums were collected and kept at -20°C until analyzed.
An established HPLC method was used to measure the glibenclamide serum concentration (12). Briefly, the dried residue of the benzene extract obtained from 0.4 ml of acidified serum was redissolved in mobile phase (acetonitril: 0.01 M phosphate buffer adjusted at pH 3.5 with the volume ratio of 40:60) and injected (20 μ L) into the system (Cecil, UK). The mobile phase was delivered at the flow rate of 1 ml/min and separation was achieved on a C18 column (4.6∞250 mm, 5 μ , Hichrom LTD, UK). Glibenclamide was detected at 225 nm wavelength with an ultraviolet detector. Chromatographic data was analyzed using Data Control program supplied with the HPLC system. A linear calibration plot prepared using peak height of the standard solutions (0.25-12 μ g/ml) of glibenclamide was used to assay the concentration of the drug in the serum samples. The mean glibenclamide serum concentrations at different times for three different treatments were compared according to one-way ANOVA using the Student-Newman-Keuls method (13).
Serum glucose concentration
Twelve male rabbits, 2.5±0.6 kg, other than those used for glibenclamide serum concentration studies were subjected to the same circumstances and dosing schedule outlined above and their serum glucose concentrations were assayed based on the standard glucose oxidase method (14) using a commercial kit according to the supplied instruction. Briefly, to 20 ml of serum sample was added required reagents and enzyme and the mixture was vortexed and incubated at 37°Cfor 35 minutes. The intensity of the developed pink color was measured spectrometrically at 500 nm against a blank solution and the concentration of glucose was determined using a linear calibration plot. The same statistical analysis mentioned above was used to compare mean serum glucose concentrations at different time for various formulations.
ResultS
Dissolution rate
The dissolution curves of pure glibenclamide, its physical mixture with the carrier Avicel PH102, and different SD systems containing the carriers Avicels PH102 and RC-591 are given in Figure 1.
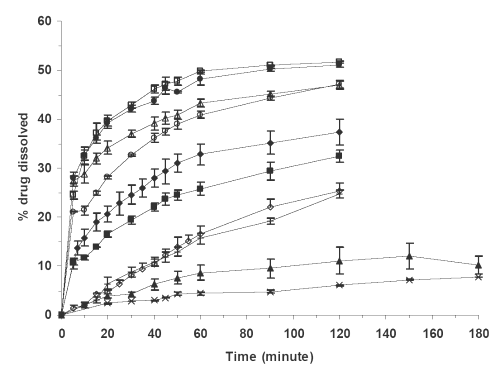
Figure 1: Percent glibenclamide dissolved in phosphate buffer dissolution medium at pH 7.25 vs. time for (x) PD, (▲) PM, (¨) SD4, (□) SD8, (◊) SD2, (○) SD5, (+) SD1, (Δ) SD6, (■) SD3, (●) SD7. Each point is the mean of three determinations. The meanings of codes are given in Table 1. The vertical bars represent the standard error of mean.
A model-independent parameter, the dissolution efficiency (DEt), was employed to compare the dissolution profiles of different formulations (15). DET was calculated according to equation 1:
(1)
Where yt is percent of drug dissolved at any time t, y100 denotes 100% dissolution, and the integral represents the area under dissolution curve between time zero and T. The time T in this study was 120 minutes.
The calculated DE120´ values for different formulations in an ascending order are shown in Table 1.
Drug serum concentrations
The arithmetic mean serum concentrations of glibenclamide against time for pure drug, physical mixture of drug and Avicel PH102 (1:19) and its SD (1:19) are seen in Figure 2.
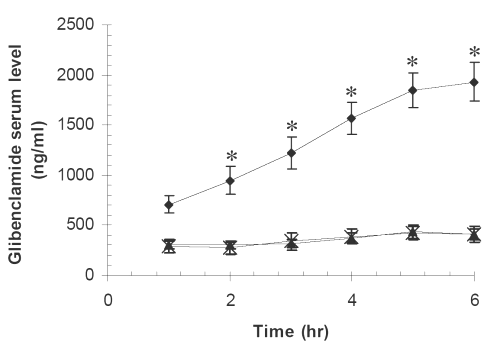
Figure 2: Glibenclamide mean serum concentrations vs. time in ten rabbits for (x) PD, (▲) PM, (●) SD7. The meanings of codes are given in Table 1. The vertical bars represent the standard error of mean. Glibenclamide serum concentrations for SD at times 2-6 hr were significantly higher than those for pure drug and the physical mixture (*P<0.001).
Serum glucose concentrations
The effects of pure glibenclamide and its PM and SD formulations on serum glucose concentrations at different time post-administration are seen in Figure 3.
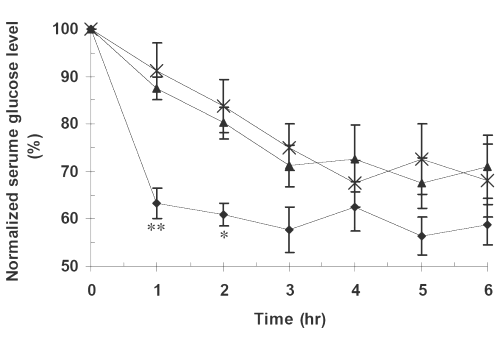
Figure 3: Mean serum glucose concentrations vs. time in twelve rabbits for (x) PD, (▲) PM, (•) SD7. The meanings of codes are given in Table 1. The concentrations are the normalized serum glucose concentrations with respect to zero time concentration and expressed as percentages. The vertical bars represent the standard error of mean. Serum glucose concentrations for SD at times 1 and 2 hr were significantly lower than those for pure drug and the physical mixture (** P<0.01, *P<0.05).
Discussion
It is evident from Figure 1 and the values of DE120´ in Table 1 that the dissolution of glibenclamide is highly dependent on its formulations. The pure drug has the lowest and the SD preparations with drug to carrier ratio of 1:19 have the highest dissolution rates. The dissolution profiles of other formulations are between these two extremes. The higher dissolution rate of PM relative to pure drug is probably due to adsorption of the hydrophilic colloidal particles of microcrystalline cellulose onto the hydrophobic glibenclamide particles, which in turn might enhance the wettability of the latter particles (7,16). The drug dissolution is increased considerably from the SD formulations and this increase strongly depends on the ratio of drug to carrier regardless of the type of Avicel used. For example, the DE120´ value is increased nearly three times by increasing the amount of carrier from one part to nineteen parts in the SD formulations. The possible causes for the enhanced dissolution from the SDs are increased wettability of the drug by the carrier, drug particle size reduction in the course of the solid dispersion preparation, polymorphic transformation of drug crystals and chemical interactions between drug and carrier (17).
Infrared spectroscopy and x-ray diffraction of glibenclamide powder and glibenclamide obtained after evaporation of its solution in chloroform show no changes in the drug molecule and crystal (Figure 4).
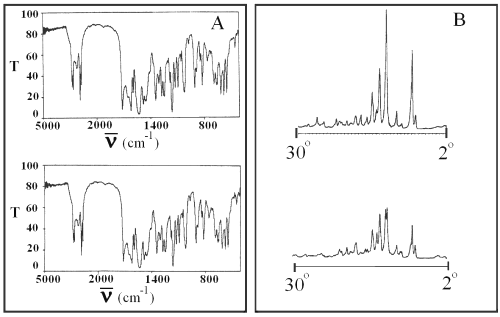
Figure 4: Infrared spectra (panel A) and x-ray diffractogram (panel B) of pure glibenclamide (top) and glibenclamide obtained after evaporation of its solution in chloroform (bottom). T is transmittance and ν is the wave number.
No changes in the IR and x-ray patterns of SD and PM formulations were observed (Figure 5).
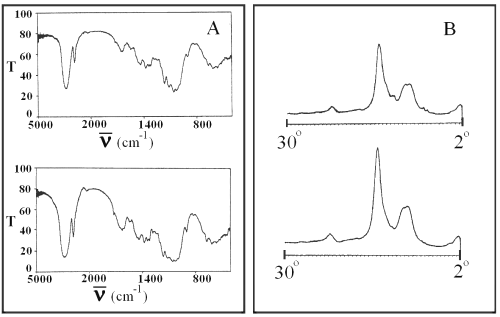
Figure 5: Infrared spectra (panel A) and x-ray diffractogram (panel B) of PM (top) and SD7 (bottom). T is transmittance and ν is the wave number.
These observations indicate that the enhanced dissolution for SD formulations is due to the increase of effective surface area of glibenclamide because of the reduction of its particle size as well as an increase of its wettability by the carriers.
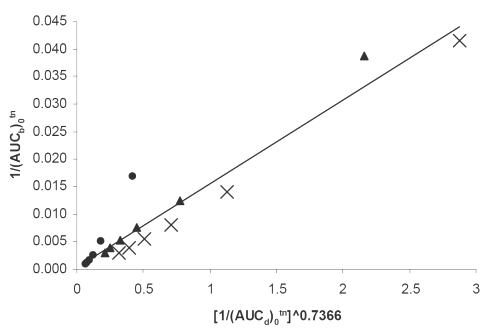
Figure 6: Double reciprocal linear plot of (AUCb)0tn vs. (AUCd)0tn ^0.7366 indicating a direct in vivo-in vitro correlation between serum drug concentrations and dissolution for (x) PD, (▲) PM, (•) SD7 formulations. See text for the meanings of (AUCb)0tn and(AUCd)0tn.
PM and PD together with one of the SD formulations exhibiting the highest dissolution rate i.e., SD7 were subjected to further in vivo studies. As it is evident from the statistical analysis the glibenclamide, serum concentrations from SD7 are significantly higher than those of PD and PM, whereas, there is no such difference between the latter two preparations (Figure 2). The difference could be explained by corresponding difference in the dissolution profiles, that is, the higher the dissolution rate, the higher the drug serum concentration. Similar results have been reported by others (3,4,7,9) involving glibenclamide solid dispersions. The higher dissolution rate can also be accounted for the higher hypoglycemic effect of SD7 formulation (Figure 3).
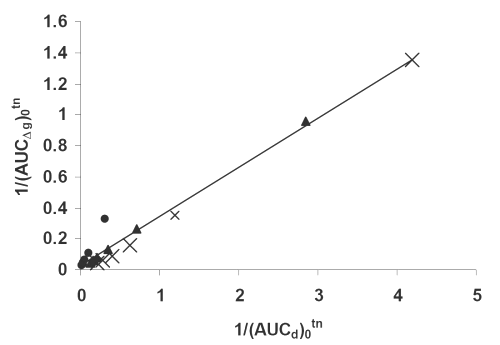
Figure 7: Double reciprocal linear plot of (AUCΔg)0tn vs. (AUCd)0tn indicating a direct correlation between hypoglycemic effect and dissolution for (x) PD, (▲) PM, (•) SD7 formulations. See text for the meanings of (AUCΔg)0tn and(AUCd)0tn.
In order to establish a reliable quantitative correlation between the in vivo and in vitro parameters of the drug, a Hill type equation was used in the form of the following double reciprocal linear relationships:
(2)
(3)
(4)
where (AUCd)0tn, (AUCb)0tn and (AUCΔg) 0tn are the areas under the drug dissolution, serum concentrations and percent reduction of serum glucose concentrations relative to its zero time concentration vs. normalized time, tn, curves between zero and tn. The normalized time is the ratio of any in vivo and in vitro sampling time with respect to the corresponding last sampling time. The normalization of time is necessary to bring in vivo and in vitro times to the same scale. In the case of PD and PM the in vivo and in vitro normalized times can directly be found from the corresponding curves, however, for the SD formulation, the normalized in vitro times corresponding to in vivo normalized time are calculated via interpolation using the dissolution curve. N is the number of data and r2 is coefficient of determination.
As it can be seen from equations 2-4, and the corresponding figures 6-8, there are excellent correlations between in vivo and in vitro parameters. These correlations indicate that both drug serum concentrations and its hypoglycemic effects are highly controlled by the drug dissolution. The higher the dissolution the higher is the corresponding in vivo parameter. The powers of the independent variables in the equations, which are obtained using iteration method, depend on kind of dependent variables used. The usefulness of such quantitative in vivo and in vitro correlations is self evident, because they could be used to predict in vivo parameters (such as areas under the drug serum concentration and hypoglycemic effect curves). In conclusion the solvent deposition technique can be employed as a reliable method for enhancing glibenclamide dissolution, serum concentrations and hypoglycemic effect. In addition, the quantitative correlations established between its in vivo and in vitro parameters can be fruitful as a simple approach for the predictive purposes.
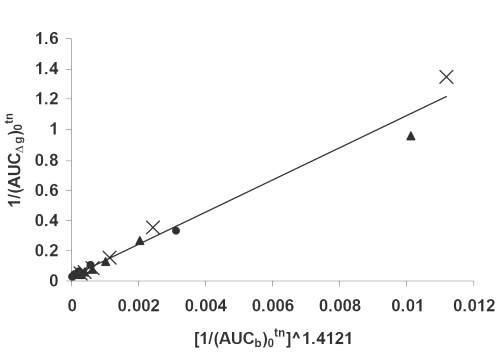
Figure 8: Double reciprocal linear plot of (AUCΔg)0tn vs. (AUCb)0tn ^1.4121 indicating a direct correlation between hypoglycemic effect and drug serum concentrations for (x) PD, (▲) PM, (•) SD7 formulations. See text for the meanings of (AUCΔg)0tn and(AUCd)0tn.
References
Sweetman, S.C., Martindale: The complete drug reference, Pharmaceutical Press, London, 2002.
Balan G, T.P., Greene DS, Marathe PH. In-vitro in-vivo correlation models for glibenclamide after administration of metformin/glibenclamide tablets to healthy human volunteers. J Pharm Pharmacol, 52: 831-838, 2000.
Deshpande, A.V., Lauwo, J.A. and Oyi, A.E. Formulation and in vitro dissolution rate studies of glibenclamide solid dispersions. Pharmacy World Journal, 46: 5153, 1990.
Ghosh, L.K., Thakur, R.S., Sharma, P.K., Ghosh, N.C. and Gupta, B.K. Development and evaluation of an ideal formulation of glibenclamide by solid dispersion techniques. Boll Chim Farm, 137: 26-29, 1998.
Kumar, R., Gupta, R.B. and Betageri, G.V. Formulation, characterization, and in vitro release of glyburide from proliposomal beads. Drug Delivery, 8: 25-27, 2001.
Lobenberg, R., Kramer, J., Shah, V.P., Amidon, G.L. and Dressman, J.B. Dissolution testing as a prognostic tool for oral drug absorption: dissolution behavior of glibenclamide. Pharm Res, 17: 439-444, 2000.
Tashtoush, B.M., Al-Qashi, Z.S. and Najib, N.M. In vitro and in vivo evaluation of glibenclamide in solid dispersion systems. Drug Dev Ind Pharm, 30: 601-607, 2004.
Valleri, M., Mura, P., Maestrelli, F., Cirri, M. and Ballerini, R. Development and evaluation of glyburide fast dissolving tablets using solid dispersion technique. Drug Dev Ind Pharm, 30: 525-534, 2004.
Varma, M.M., Jayaswal, S.B. and Singh, J. In-vitro and in-vivo evaluation of fast release solid dispersions of glibenclamide. Indian Drugs, 29: 608-611, 1992.
van der Watt, J.G., Parrot, E.L. and Devilliers, M.M. A comparison of interaction and solvent deposition mixing. Drug Dev Ind Pharm, 22: 741-746, 1996.
Yen, S.Y., Chen, C.R., Lee, M.T. and Chen, L.C. Investigation of dissolution enhancement of nifedipine by decomposition on superdisintigrants. Drug Dev Ind Pharm, 23: 313-317, 1997.
Emilsson, H., Sjoberg, S. Svender, M. and Christenson, I. High performance liquid chromatographic determination of glibenclamide in human plasma and urine. J Chromatogr, 383: 93-102, 1986.
Armitage, P., Statistical methods in medical research, Blackwell Scientific Publications, London, 1977.
El-Sayed, Y.M., Suleiman, M.S., Hasan, M.M., Abdel-Hamid, M.E., Najib, N.M., Sallam, E.S. and Shubair, M.S. Comparison of the pharmacokinetics and pharmacodynamics of two commercial products containing glibenclamide. Int J Clin Pharmacol Ther Toxicol, 27: 551-557, 1989.
Khan, K.A. The concept of dissolution efficiency. J Pharm Pharmacol, 27: 48-49, 1975.
Barzegar-Jalali, M., Maleki, N., Garjani, A., Khandar, A.A., Haji-Hosseinloo, M., Jabbari, R., Dastmalchi, S. Enhancement of dissolution rate and anti-inflammatory effects of piroxicam using solvent deposition technique. Drug Dev Ind Pharm, 28: 681-686, 2002.
Leuner, C. and Dressman, J. Improving drug solubility for oral delivery using solid dispersions. Eur J Pharm Biopharm, 50: 47-60, 2000.
Corresponding Author: Mohammad Barzegar-Jalali, Department of Pharmaceutics, School of Pharmacy, Tabriz University of Medical Sciences, Tabriz, 51664, Iran. barzegar_jalali@yahoo.com
Published by the Canadian Society for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences.
http://www.cspscanada.org
