J Pharm Pharmaceut Sci (www.cspscanada.org) 8(3):565-577, 2005
Aminopyrimidinimino isatin analogues: Design of novel non- nucleoside HIV-1 reverse transcriptase inhibitors with broad-spectrum chemotherapeutic properties.
Dharmarajan Sriram, Tanushree Ratan Bal, Perumal Yogeeswari
Medicinal
Chemistry Research Laboratory,
Pharmacy
Group, Birla Institute of Technology
and Science, Pilani 333031,
Received April 11, 2005; Revised September 22, 2005; Accepted October 19, 2005; Published October 20, 2005
Corresponding
Author:
Dharmarajan Sriram, Medicinal Chemistry
Research Laboratory, Pharmacy group, Birla Institute of Technology and Science,
Pilani 333031,
ABSTRACT: Purpose: HIV is the most significant risk factor for many
opportunistic infections such as tuberculosis, hepatitis, bacterial infections and
others. In this paper, we describe an aminopyrimidinimino
isatin lead compound as a novel non-nucleoside reverse transcriptase
inhibitor with broad-spectrum chemotherapeutic properties for the effective
treatment of AIDS and AIDS-related opportunistic infections. Methods: The
synthesis of various aminopyrimidinimino isatin derivatives was
achieved in two steps and evaluated for anti-HIV, anti-HCV, antimycobacterial
and antibacterial activities. Results: Compound
1-cyclopropyl-6-fluoro-1,4-dihydro-4-oxo-7[[N4-[3’-(4’-amino-5’-trimethoxybenzylpyrimidin-2’-yl)imino-1’-isatinyl]
methyl]N1-piperazinyl]-3-quinoline carboxylic acid (14) emerged as the most potent
broad-spectrum chemotherapeutic agent active against HIV, HCV, M.
tuberculosis and various pathogenic bacteria. Among the synthesized compounds compound 14 and 15 emerged as more
promising broad-spectrum chemotherapeutic agents.
Introduction
Acquired immunodeficiency syndrome (AIDS) is caused by the retrovirus, human immunodeficiency virus (HIV) [1]. The HIV infection, which targets the monocytes expressing surface CD4 receptors, eventually produces profound defects in cell-mediated immunity [2]. Over time infection leads to severe depletion of CD4 T-lymphocytes (T-cells) resulting in opportunistic infections (OIs) like tuberculosis (TB), fungal, viral, protozoal and neoplastic diseases and ultimately death. TB is the most common OI in people with AIDS and it is the leading killer of people with AIDS. The co-infection by hepatitis C virus (HCV) and HIV is quite common, mainly because these infections share the same parenteral, sexual and vertical routes of transmission [3]. Although classical OIs are now rarely seen, the toxicity of antiretroviral drugs as well as liver diseases caused by HCV represents an increasing cause of morbidity and mortality among HIV-positive persons. Predisposing liver damage favors a higher rate of hepatotoxicity of antiretroviral drugs, which can limit the benefit of HIV treatment in some individuals [4]. Through logic and orderly thinking, it appears that an ideal drug for HIV/AIDS patients should suppress HIV replication thereby acting as an anti-HIV drug and also should treat OIs like TB, hepatitis and other bacterial infections. Earlier work in our laboratory has identified various isatinimino derivatives exhibiting broad-spectrum chemotherapeutic properties [5]. As a continuation of our effort in developing broad-spectrum chemotherapeutics, we undertook the present study to design, synthesize and evaluate aminopyrimidinimino isatin analogues, which could suppress HIV replication and also inhibit the opportunistic microorganisms.
Experimental
Chemistry
Melting points were determined in one end open capillary tubes on a Büchi 530 melting point apparatus and are uncorrected. A domestic microwave oven with the following specifications had been used : Make LG; Input 220V~50 Hz, 980 W, 4.7 A; Frequency 2450 MHz. Infrared (IR), proton nuclear magnetic resonance (1H-NMR) spectra and 13C NMR were recorded for the compounds on Jasco IR Report 100 (KBr) and Brucker Avance (300MHz) instruments and Varian unity 400 (50MHz) spectrometer respectively. Chemical shifts are reported in parts per million (ppm) using tetramethyl silane (TMS) as an internal standard. Elemental analyses (C, H, and N) were undertaken with Perkin-Elmer model 240C analyzer. The homogeneity of the compounds was monitored by ascending thin layer chromatography (TLC) on silicagel-G (Merck) coated aluminium plates, visualized by iodine vapour. Developing solvents were chloroform-methanol (9:1). The pharmacophoric distance map and log P values were determined using Alchemy-2000 and Scilog P software (Tripos Co.).
Synthesis
of (3-{[4’-amino-5(3’’, 4’’, 5’’- trimethoxybenzyl) pyrimidin-2’-yl]}
imino}-5-bromo-1, 3-dihydro-2H-indol-2-one)
Equimolar quantities (0.01 mole)
of 5-bromoisatin and 5-(3’, 4’, 5’-trimethoxybenzyl)-two, 4-diaminopyrimidine
(Trimethoprim) were dissolved in warm ethanol containing 1 ml of glacial acetic
acid. The reaction mixture was irradiated in an unmodified
domestic microwave oven [17] at 80% intensity with 30 sec/cycle for 3 minutes
and set aside. The resultant solid was washed
with dilute ethanol dried and recrystallized from ethanol-chloroform mixture.
Yield 84.2%; m.p.: 185 °C; IR (KBr) : 3300, 2040,
1660, 1620,1580 cm-1; 1H-NMR (CDCl3) δ
(ppm): 3.18 (s, 2H, CH2), 3.7 (s, 9H, -OCH3), 5.6 (s, 2H,
NH2), 6.7-7.2 (m, 6H, Ar-H), 10.7(s, 1H, -NH).
General
procedure for the preparation of Mannich bases
To a suspension of
3-{[4’-amino-5-(3’’,4’’,5’’- trimethoxybenzyl) pyrimidin-2’-yl]} imino}-5-bromo-1,3-dihydro-2H-indol-2-one
(0.02mol) in ethanol was added appropriate secondary amines (0.02 mole) and 37%
formaldehyde (0.5 ml) and irradiated in a microwave oven at an intensity of 80%
with 30sec/cycle. The number of cycle in turn depended on the completion of the
reaction, which was checked by TLC. The reaction
timing varied from 1.5-3 min. The solution obtained after the completion of the
reaction was kept at 0oC for 30 min and the
resulting precipitate was recrystallized from a mixture of DMF and water.
(3-{[4’-amino-5’-(3’’,4’’,5’’-
trimethoxybenzyl) pyrimidin-2’-yl]} imino}-5-bromo-1-[( diethylamino) methyl]-1,3-dihydro-2H-indol-2-one) (2)
Yield: 70.26%;
m.p.: 68oC ; IR (KBr) : 3010, 2850, 2840, 1730, 1616, 1506, 1236,
1129 cm-1; 1H-NMR (CDCl3) δ (ppm): 1.8
(t, 6H, CH3 of C2H5, J = 15 Hz), 3.16 (s, 2H,
CH2 of benzyl), 3.7 (s, 9H, -OCH3), 4.2 (q, 4H, CH2
of C2H5, J = 12 Hz), 5.1 (s, 2H, -NCH2N-), 5.6
(s, 2H, NH2), 6.8-7.26 (m, 6H, Ar-H) ; 13C NMR (DMSO-d6)
δ (ppm): 13.0 (2C, CH3s of C2H5), 41.3 (CH2),
46.9 (2C, CH2s of C2H5), 56.1 (2C, OCH3s
at 3- and 5- positions), 56.4 (OCH3 at 4-position), 70.1 (CH2),
106.3 (2C at 2- and 6- positions of trimethoxyphenyl), 114.2 (C at 5-position
of pyrimidine), 118.8 (C at 5-position of indole), 119.9 (C at 9-position of
indole), 123.9 (C at 7-position of indole), 130.5 (C at 1-position of trimethoxyphenyl),
132.9 (C at 4-position of indole), 134.1 (C at 6-position of indole), 136.2 (C
at 4-position of trimethoxyphenyl), 146.4 (C at 8-position of indole), 150.7 (2C
at 3- and 5-positions of trimethoxyphenyl), 161.1 (C at 4-position of
pyrimidine), 163.0 (C at 6-postion of pyrimidine), 163.2 (C at 3-position of
indole),163.5 (C at 2-position of indole), 164.1 (C at 2-position of
pyrimidine); Calculated for C27H31N6O4Br
: C, 55.58; H, 5.36; N, 14.4; found: C, 55.48; H, 5.39; N, 14.60.
(3-{[4’-amino-5’-(3’’,4’’,5’’-
trimethoxybenzyl) pyrimidin-2’-yl]} imino}-5-bromo-1-[( 4-benzyl piperazinyl) methyl]-1,3-dihydro-2H-indol-2-one)
(3)
Yield: 75.10%;
m.p.: 87oC ; IR (KBr) : 3010, 2850, 2840, 1730, 1616, 1500, 1240, cm
; 1H-NMR (CDCl3) δ (ppm): 3.17 (s, 2H, CH2 of
trimethoxybenzyl), 3.65 (s, 9H, -OCH3), 3.9 -4.1 (m, 8H,
piperazine-H), 4.36 (s, 2H, CH2 of benzylpiperazine), 5.2 (s, 2H,
-NCH2N-), 5.65 (s, 2H, NH2), 6.67-7.82 (m, 11H, Ar-H) ;
13C NMR (DMSO-d6) δ (ppm): 41.3 (CH2), 50.3
(2C of 2- and 6-positions of piperazine), 52.2 (2C of 3- and 5-positions of
piperazine), 56.1 (2C of OCH3s at 3- and 5-positions), 56.4 (C of OCH3 at 4-position),
60.1 (CH2) 70.1 (CH2), 106.3 (C at 2-and 6-positions of
trimethoxyphenyl), 114.2 (C at 5- position of pyrimidine), 118.8 (C at
5-position of indole), 119.9 (C at 9-position of indole), 123.9 (C at
7-position of indole), 127.3 (C at 4- position of phenyl), 128.5 (2C at 3- and
5- positions of phenyl), 128.8 (2C at 2- and 6- positions of phenyl), 130.5 (C
at 1-position of trimethoxyphenyl), 132.9 (C at 4-position of indole), 134.1 (C
at 6-position of indole), 135.5 (C at 1-position of phenyl), 136.2 (C at
4-position of trimethoxyphenyl), 146.4 (C at 8-position of indole), 150.7 (C at
3- and 5-positions of trimethoxyphenyl), 161.1 (C at 4- position of pyrimidine),
163.0 (C at 6- position of pyrimidine), 163.2 (C at 3-position of indole), 163.5
(C at 2-position of indole), 164.1 (C at 2-position of pyrimidine) Calculated for C34H36N7O4Br
: C, 59.48; H, 5.28; N, 14.28; found: C, 59.60; H, 5.20; N, 14.32.
(3-{[4’-Amino-5’-(3’’,4’’,5’’-
trimethoxybenzyl) pyrimidin-2’-yl] imino}-5-bromo-1-[( 3-chlorophenyl
piperazinyl) methyl]-1,3-dihydro-2H-indol-2-one) (4)
Yield: 62.82%;
m.p.: 84oC ; IR (KBr) : 3010, 2850, 2830, 1730, 1620, 1500, 1240, cm
; 1H-NMR (CDCl3) δ (ppm): 3.17 (s, 2H, CH2 of
trimethoxybenzyl), 3.65 (s, 9H, -OCH3), 3.9 -4.1 (m, 8H,
piperazine-H), 5.2 (s, 2H, -NCH2N-), 5.65 (s, 2H, NH2),
6.67-7.82 (m, 10H, Ar-H) ; Calculated for C33H33N7O4ClBr
: C, 56.06; H, 4.7; N, 13.87; found: C, 56.12; H, 4.67; N, 13.62
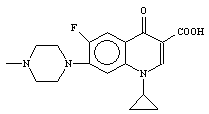
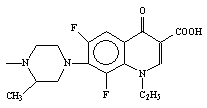
1-cyclopropyl-6-fluoro-1,4-dihydro-4-oxo-7-[[N4-[3’-(4’-amino-5’- trimethoxybenzyl
pyrimidin-2’-yl)imino-1’-(5-bromoisatinyl)] methyl] N’-piperazinyl]-3-quinoline
carboxylic acid (14)
Yield: 76%;
m.p.: 222o C ; IR (KBr) : 3010, 2850, 2840, 1736, 1620, 1506, 1236,
1125 cm ; 1H-NMR (CDCl3) δ (ppm): 0.88-1.1 (m, 4H,
cyclopropyl-H), 3.3 (s, 2H, CH2 of benzyl), 3.5 (m, 1H,
cyclopropyl-H ), 3.62 (s, 9H, -OCH3), 3.7-4.1 (m, 8H,
-piperazine-H), 5.1 (s, 2H, -NCH2N), 5.8 (s, 2H, NH2),
6.58-8.60 (m, 9H, Ar-H), 8.6 (s, 1H, C2-H) ; 13C NMR
(DMSO-d6) δ (ppm): 5.6 (C at 2- and 3-positions of cyclopropyl),
36.0 (C at 1-position of cyclopropyl), 41.3 (CH2), 49.6 (2C of 3-
and 5- positions of piperazine), 49.9 (2C of 2- and 6-positions of piperazine),
56.3 (2C od OCH3s at 3- and 5-positions), 56.6 (C of OCH3
at 4-position), 70.1 (CH2), 100.0 (C at 8-position of quinoline),
106.3 (2C at 2- and 6-positions of trimethoxyphenyl), 109.3 (C at 3-position of
quinoline), 114.2 (C at 5-position of pyrimidine), 116.4 (C at 5-position of
quinoline), 118.2 (C at 10-position of quinoline), 118.8 (C at 5-position of
indole), 119.9 (C at 9-position of indole), 123.9 (C at 7-position of indole),
130.5 (C at 1-position of trimethoxyphenyl), 132.9 (C at 4-position of indole),
134.1 (C at 6-position of indole), 136.2
(C at 4-position of trimethoxyphenyl), 140.5 (C at 9-position of quinoline),
143.9 (C at 7-position of quinoline), 144.6 (C at 6-position of quinoline), 146.4
(C at 8-position of indole), 148.0 (C at 2-position of quinoline), 150.7 (C at
3- and 5-positions of trimethoxyphenyl), 161.1 (C at 6-position of pyrimidine),
163.0 (C at 4-position of pyrimidine), 163.2 (C at 3-position of indole), 163.5
(C at 2-position of indole), 164.1 (C at 2-position of pyrimidine), 166.2
(COOH) , 177.4 (C at 4-position of quinoline); Calculated for C40H38N6O7FBr
: C, 57.08; H, 4.55; N, 13.31; found: C, 57.12; H, 4.61; N, 13.30.
Anti-HIV activity
Candidate agents were dissolved in dimethylsulfoxide, and then diluted 1:100
in cell culture medium before preparing serial half- log10 dilutions. T4
lymphocytes (CEM cell-line) were added and after a brief interval
HIV-1 was added, resulting in a 1:200 final dilution of the compound.
Uninfected cells with the compound served as a toxicity control, and infected
and uninfected cells without the compound served as basic controls. Cultures were incubated at 37˚C in a 5% carbon dioxide
atmosphere for 6 days. The tetrazolium salt, XTT {2,3-bis
(2-methoxy-4-nitro-5-sulfophenyl)-5-[(phenylamino) carbonyl]-2H-tetrazolium
hydroxide} was added to all the wells, and cultures were incubated to allow formazan
color development by viable cells. Individual wells were
analyzed spectrophotometrically to quantitative formazan production, and
in addition were viewed microscopically for detection of viable cells and
confirmation of protective activity.
HIV-1RT assay
The reaction mixture (50µl) contained 50mM Tris–HCl (pH 7.8), 5mM dithiothreitol, 30 mM glutathione, 50 µM EDTA, 150 mM KCl, 5 mM MgCl2, 1.25 µg of bovine serum albumin, an appropriate concentration of the radiolabeled substrate [3H] dGTP, 0.1 mM poly(νC)·oligo(dG) as the template/primer, 0.06% Triton X-100, 10 µl of inhibitor solution (containing various concentrations of compounds), and 1 µl of RT preparation. The reaction mixtures were incubated at 37°C for 15 min, at which time 100 µl of calf thymus DNA (150 µg/ml), 2 ml of Na4P2O7 (0.1 M in 1 M HCl), and 2 ml of trichloroacetic acid (10% v/v) were added. The solutions were kept on ice for 30 min, after which the acid-insoluble material was washed and analyzed for radioactivity. For the experiments in which 50% inhibitory concentration (IC50) of the test compounds was determined, fixed concentration of 2.5µM [3H] dGTP was used.
Antiviral and
cytotoxicity assays for HCV
Cell culture
Huh-7 cells the subgenomic HCV replicon BM4-5 cells were
maintained in Dulbecco0s modified Eagle’s medium (DMEM) (Life Technologies)
supplemented with 10% fetal bovine serum, 1% l-glutamine, 1% l-pyruvate, 1%
penicillin and 1% streptomycin supplemented with 500 mg/mL G418 (Geneticin,
Invitrogen). Cells were passaged every 4 days.
Cytotoxicity
Huh-7 cells were respectively seeded
at a density of 3 X10-4 cells/well in 96-well plates for the
cell-viability assay, or at a density of 6X10-5 cells/well in
six-well plates for the antiviral assay. Sixteen hours post seeding, cells were treated with the compounds at 50μg/mL for 3 days.
The administration of each drug was renewed each day.
Other drugs, including ribavirin (ICN Pharmaceuticals,
Antiviral assay
Total tRNA (transfer RNA) was extracted
from six-well plates with the ‘Extract All’ reagent (Eurobio), which is a
mixture of guanidinium thiocyanate–phenol–chloroform. Northern Blot analysis
was then performed using the NorthernMaxTM-Gly (Ambion) kit, following
manufacturer’s instruction. Ten micrograms of tRNA was
denatured in glyoxal buffer at 50˚C for 30 min and separated by agarose
gel electrophoresis, then transferred for 12 h onto a charged nylon membrane
(Biodyne B, Merck Eurolab). Hybridization was carried
out with three different [32P] CTP-labeled riboprobes
obtained by in-vitro transcription (Promega). These probes were
complementary to the NS5A region of the HCV genome, and to the cellular gene
GAPDH, respectively. First, the blot was hybridized
with two riboprobes directed against the negative strand of HCV RNA and the
GAPDH mRNA, respectively. After one night of hybridization at 68˚C, the
membrane was washed then exposed to X-ray film and a phosphor
screen for quantitative analysis. The amount of GAPDH mRNA was used as an internal loading control to standardize the
amount of HCV RNA detected. The same membrane was
subsequently hybridized with a negative-sense riboprobe to determine the
level of HCV-positive strand RNA using the same approach.
Antimycobacterial activity
Primary screening was conducted at
6.25 μg/ml against Mycobacterium tuberculosis strain H37Rv
(ATCC 27294) in BACTEC 12B medium using a broth microdilution assay, the
microplate Alamar Blue Assay (MABA) .
In vitro antibacterial activity
Compounds were evaluated for their
in-vitro antibacterial activity against 28 pathogenic bacteria procured
from the Department of Microbiology,
In vivo antibacterial activity (mouse
protection test)
The in-vivo antibacterial activity of the test compounds was determined in CF-strain male mice (20–25 g body weight, six per group). The protocol is approved by Institute Animal Ethical Committee (IAEC/RES/11). The mice were infected intraperitoneally with a suspension containing an amount of the indicated organism slightly greater than its lethal dose 100 (LD100). The mice were treated orally (p.o.) with a specific amount of the test compound administered at 1 and 4 h after infection. ED50 values were calculated by interpolation among survival rates in each group after a week. They express the total dose of compound (mg/kg) required to protect 50% of the mice from an experimentally induced lethal systemic infection of the indicated organism.
RESULTS AND
DISCUSSIONS
Design
To qualify as a non-nucleoside
reverse transcriptase inhibitors (NNRTI), the compound should interact
specifically with a non-substrate binding site of the reverse transcriptase
(RT) of HIV-1, and inhibit the replication of HIV-1 at a concentration that is
significantly lower than the concentration required affecting normal cell
viability [6]. Based on these premises, more than thirty different classes of
NNRTIs could be considered [7]. Several
studies have revealed that although the NNRTIs seemingly belong to a widely
diverging classes of compounds, but on closer inspection it has been elucidated
that most of them possess some features in common, that is a (thio)
carboxamide, (thio) acetamide or (thio) urea entity (‘body’) which is
hydrophilic in nature, surrounded by two hydrophobic, mostly aryl moieties
(‘wings’), one of which is quite often substituted by a halogen group.
Thus, the overall structure may be considered
reminiscent of a butterfly with hydrophilic centre (‘body’) and two hydrophobic
outskirts (‘wings’). The ‘butterfly-like’ conformation has
been proven by crystallographic analysis for Nevirapine [8] and TBZ.
Based on this hypothesis, a 3D-pharmacophoric distance model was
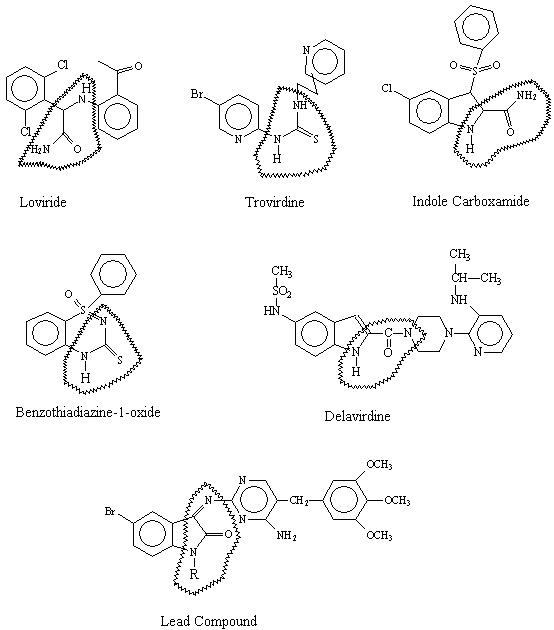
derived utilizing eight well-known NNRTIs, i.e. Nevirapine, Delavirdine,
Efavirenz, Trovirdine, Loviride, indole carboxamide, benzothiadiazine-1-oxide
and thiocarboxanilide. All the ligands were geometrically
optimized based on the internal strain energy calculated by molecular
mechanics calculations (MM3 parameterization) in Alchemy Tripos software to
ensure uniform sampling of low energy conformers. Then the essential structural
components like atoms, centroids of collection of atoms, electron lone pair
positions, steric and electrostatic potentials etc were
matched in the three-dimensional space of the energetically accessible
conformations of the ligands, to arrive at the 3-point pharmacophore model
proposed below (Fig 2).
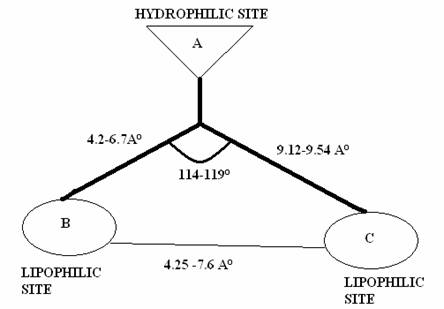
Figure 2: Schematic representation of a butterfly-like
configuration of NNRTI’S and the pharmacophoric
distance map
In the present study, the
aminopyrimidinimino isatin analogues are designed in accord
with this hypothesis. The iminocarbamoyl moiety (-N=C-CO-N-) constitutes the
‘body’ and the aryl ring of isatin and the pyrimidine derivative constitute the
‘wings’ as depicted in
Fig.1. The crucial structural components included in the
proposed model contain a hydrophilic centre (A), which is
surrounded by 2 hydrophobic outskirts denoted by B and C. The distance
between the 3-pharmacophoric points were calculated
for minimum four different conformations and are represented as mean standard
deviation. The lead compound was found to comply
within the specification of the pharmacophoric distance map (Fig. 2 and
Table
1). During the development of this 3D-pharmacophoric model, molecular
superposition techniques have also been used to investigate similarities and
differences between the selected points in the test molecule
(aminopyrimidinimino isatin analogue) and the corresponding points in the
reference molecule (Nevirapine, Efavirenz, and Delavirdine) calculated by means
of RMS (Root mean square deviation)
value. It was deduced from the RMS fit value that the
structure fits appreciably with Delavirdine with RMS value of 0.075 (Fig 3)
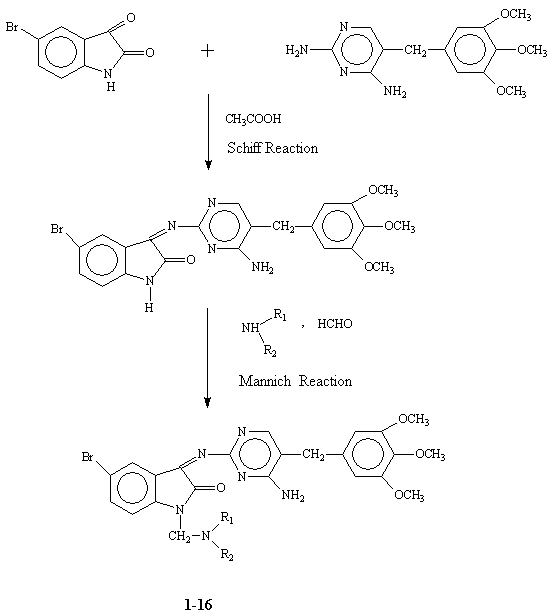
Synthesis
The synthesis of various
aminopyrimidinimino isatin derivatives was achieved in
two steps (Fig.4) [9]. 5-Bromoisatin was condensed with 5-trimethoxybenzyl-2,4-diamino pyrimidine in the presence of glacial acetic acid
to form Schiff’s base. The N-Mannich bases of the above Schiff’s base were synthesized by condensing acidic imino group of isatin
with formaldehyde and various secondary amines. All compounds (Table 2 and
3)
gave satisfactory elemental analysis. IR, 1H-NMR
and 13C spectra were consistent with the assigned structures.
Biological
activities
The synthesized compounds were evaluated for their inhibitory effect on the
replication of HIV-1 in MT-4 and CEM cell lines (Table 4) [10]. In the MT-4
cell lines, compound 12 and 15 were found to be the most active
against replication of HIV-1 with EC50 of 5.6 and 7.6 μM
respectively and their selectivity index (SI=CC50/EC50)
was found to be more than 12 with maximum protection of 94-126%. When compared
to reference standard Nevirapine (EC50 = 0.1 μM) the
synthesized compounds were less active. Other compounds (2, 3, 7,
14, and 16) showed maximum protection of 56%-88% with SI of 2-10.
In the T4 lymphocytes (CEM cell lines), the compounds showed marked anti-HIV
activity (15-48%) at a concentration below their toxicity threshold. The loss
of activity might be due to degeneration / rapid metabolism in the culture
conditions used in the screening procedure. Overall, seven compounds of the 16
new derivatives developed in this work showed inhibition against replication of
HIV-1 in MT-4 cells with EC50 ranging from 5.6-22.6 μM.
Table 1.
The distance between the pharmacophoric functional
groups of anti-HIV drugs and the lead compound
|
Compound |
AB (in Å) |
BC (in Å) |
CA (in Å) |
|||
|
Lower limit |
Upper limit |
Lower limit |
Upper limit |
Lower limit |
Upper limit |
|
|
Delavirdine |
4.328 ± 0.04 |
6.705 ± 0.15 |
4.256 ± 0.19 |
7.542 ± 0.35 |
9.156± 0.04 |
9.382± 0.04 |
|
Trovirdine |
4.235 ± 0.01 |
6.635 ± 0.02 |
4.289 ± 0.08 |
7.269 ± 0.16 |
9.168± 0.04 |
9.426± 0.04 |
|
Loviride |
4.356 ± 0.03 |
6.709 ± 0.12 |
4.254 ± 0.06 |
7.129 ± 0.14 |
9.132± 0.04 |
9.368± 0.04 |
|
Indole carboxamide |
4.359 ± 0.01 |
6.705 ± 0.20 |
4.562 ± 0.14 |
7.478 ± 0.07 |
9.125± 0.04 |
9.434± 0.04 |
|
Efavirenz |
4.425 ± 0.05 |
6.689 ± 0.16 |
4.247 ± 0.23 |
7.211 ± 0.21 |
9.145± 0.04 |
9.440± 0.04 |
|
Nevirapine |
3.854 ± 0.02 |
6.538 ± 0.04 |
4.268 ± 0.31 |
7.603 ± 0.02 |
9.215± 0.04 |
9.421± 0.04 |
|
Benzothiadiazine-1-oxide |
4.512 ± 0.02 |
6.459 ± 0.03 |
4.269 ± 0.14 |
7.545 ± 0.01 |
9.129± 0.04 |
9.406± 0.04 |
|
Thiocarboxanilide |
4.229 ± 0.04 |
6.523 ± 0.11 |
4.223 ± 0.07 |
7.147 ± 0.39 |
9.169± 0.04 |
9.398± 0.04 |
|
Lead Compound |
4.235 ± 0.18 |
6.459 ± 0.12 |
4.218 ± 0.09 |
7.547 ± 0.15 |
9.159± 0.01 |
9.431± 0.02 |
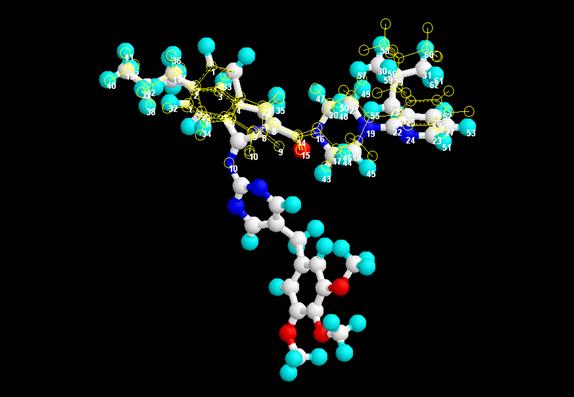
Figure: 3 Superimposition and RMS fit of the proposed lead
compound and Delavirdine (RMS value = 0.075)
Two compounds (14 and 15)
were evaluated for the inhibitory effects on HIV-1 RT
enzyme [11] and their IC50 values were found to be 28.4 ± 4.4 and 40.2 ± 8.6 μM respectively. The in-vitro IC50 values for
HIV-1 RT with Poly (νC) oligo (dG) as the template / primer were
significantly higher than the corresponding EC50 values for
inhibition of the cytopathic effect of HIV-1 in MT-4 cell culture. This
discrepancy is not unusual for NNRTI’s as it may
reflect the differences between the in
vitro HIV-1 RT assay, in which a synthetic template/primer is used, and the
cellular systems [12]. All the synthesized compounds were
also evaluated preliminarily for their inhibition of HCV viral RNA
replication in Huh-7 cells at 50-μg/ ml [13], and the results are
presented in Table 4. Among these, four compounds (10, 14, 15 and
16) were found to be less toxic to Huh-7 cells (cell growth of > 80%) and inhibited HCV viral RNA replication at about
80-86%. Two compounds (1 and 13) inhibited 100% viral replication
but they were toxic to Huh-7 cells. Summarizing, twelve compounds were active
against HCV RNA replication showing 80% inhibition at 50
μg/ml. This paper is the first of its kind in which isatin
derivatives are reported to possess anti- HCV
activity.
The synthesized compounds were
also screened against M. tuberculosis strain H37Rv (ATCC
27294) in BACTEC 12B medium initially at 6.25 μg/ml (Table 4) [14]. Three
compounds (14, 15 and 16) showed complete inhibition
(100%) of M. tuberculosis in the primary screening. In the secondary level screening the actual minimum inhibitory concentration
(MIC) and cytotoxicity in VERO cells of these three compounds were determined.
The MIC’s of these compounds were found to be 3.13
μg/ml and they were not cytotoxic up to 62.5 μg/ml to VERO cells.
All
the compounds were evaluated for their in-vitro antibacterial
activity against 24 pathogenic bacteria by conventional agar dilution
procedures [15] and the results of these assays are summarized in
Table 5 and
6.
The
data for Ciprofloxacin, Lomefloxacin and Ggatifloxacin were included for
comparison. The antibacterial activity data revealed that all the test
compounds showed mild to moderate activity against tested bacteria. The most
sensitive organisms for the tested compounds were S sonnei, Vibrio mimicus, V. flurialis, V. cholerae0139,
V. parahaemolyticus and Citrobacter ferundii as these compounds
inhibited them at a concentration less than 50 μM.
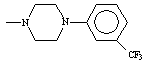
Table 2. Physical constants of the synthesized compounds 1-10
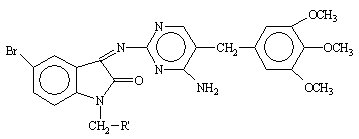
|
Compound |
R’ |
Molecular
Formula |
Molecular
Weight |
Yield
(%) |
M.P.
(°C) |
|
1 |
|
C31H39N6O4Br |
639.58 |
69.10 |
104 |
|
2 |
|
C27H31N6O4Br |
583.49 |
70.26 |
68 |
|
3 |
|
C34H36N7O4Br |
686.60 |
75.10 |
87 |
|
4 |
|
C33H33N7O4ClBr |
707.01 |
62.82 |
84 |
|
5 |
|
C28H32N7O4Br |
610.50 |
65.12 |
85 |
|
6 |
|
C34H36N7O5Br |
702.59 |
69.50 |
117 |
|
7 |
|
C34H36N7O5Br |
702.59 |
72.10 |
82 |
|
8 |
|
C34H36N7O5Br |
702.59 |
70.80 |
83 |
|
9 |
|
C33H34N7O4Br |
672.57 |
71.21 |
130 |
|
10 |
|
C32H33N8O4Br |
673.56 |
71.56 |
76 |
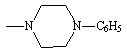
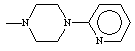
Compound 7 which contain
3-methoxyphenyl piperazinomethyl moiety at N-1 position was found to be the
most active compound that was more potent than lomefloxacin against K.
ozaenae, S. sonnei, V. flurialis, V. cholerae0139, V. parahaemolyticus,
E. coli NCTC 10418, E tarda, P mirabilis, S. typhi, S. enteritidis, C.
ferundii, enterobacter and B. megatherius.
Compound 14, containing a ciprofloxacin moiety at N-1 position was found to be more active than ciprofloxacin against 17
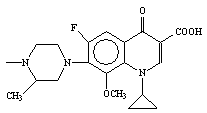
tested bacteria. When compared to Llomefloxacin, compound 15
(Lomefloxacin derivative) was found to be more active against 23 tested
bacteria. Compound 16 bearing Gatifloxacin at N-1 position was found to be more active than Gatifloxacin against 15
tested bacteria. These data are in consistent with our earlier results [16].
Table 3.
Physical constants of the synthesized compounds 11-16
|
Compound |
R’ |
Molecular
Formula |
Molecular
Weight |
Yield
(%) |
M.P.
(°C) |
|
11 |
|
C34H33N7O4F3Br |
740.57 |
66.12 |
146 |
|
12 |
|
C27H29N6O5Br |
597.46 |
65.50 |
88 |
|
13 |
|
C27H29N6O4Br |
581.46 |
64.82 |
94 |
|
14 |
|
C40H38N8O7FBr |
841.68 |
76.15 |
222 |
|
15 |
|
C40H39N8O7F2Br |
861.68 |
76.50 |
257 |
|
16 |
|
C42H42N8O8FBr |
885.73 |
75.86 |
137 |
In-vivo antibacterial activity of some selected compounds against an experimentally induced infection of mice after oral administration [16] are presented in Table 7, along with the in-vitro activity against the infecting organism E. coli NCTC 10418. Ciprofloxacin and Lomefloxacin were used as reference compounds. Compound 14 was found to be 3 times more active (ED50: 0.46 mg/kg body weight) than Ciprofloxacin (ED50: 1.25 mg/kg) while compound 15 was equally active as Lomefloxacin with ED50 of 1.87 mg/kg against the tested bacteria. Thus, four compounds showed very good activity against various pathogenic bacteria, and among the synthesized compounds, compound 14 and 15 emerged as more promising broad-spectrum chemotherapeutic agents. The more activity of the synthesized compounds might be due to the dual inhibition of bacterial enzymes dihydrofolate reductase (trimethoprim nucleus) and DNA gyrase (fluoroquinoline nucleus).
ACKNOWLEDGEMENTS
The authors acknowledge M/S
VenkarChem,
Table 4. Anti-HIV, anti-HCV and antimycobacterial activity
|
Compound |
Anti-HIV activity (µM) |
Anti-HCV |
Antimycobacterial activity at 6.25
μg/ml |
||||||
|
Cell growth (%) |
Viral RNA replication (%) |
% Inhibition |
|||||||
|
MT-4 cell line |
CEM cell line |
||||||||
|
EC50a |
CC50b |
% Protection |
EC50 a |
CC50b |
% Protection |
||||
|
1 |
>
36.1 |
36.1 |
29.6 |
NT |
NT |
NT |
11 |
100 |
68 |
|
2 |
19.2 |
62.6 |
72.6 |
>
42.8 |
42.8 |
39.93 |
84 |
76 |
46 |
|
3 |
7.8 |
79.1 |
88.6 |
>
74.0 |
74.0 |
42.40 |
62 |
74 |
NT |
|
4 |
>
41.6 |
41.0 |
29.1 |
>
37.2 |
37.2 |
29.73 |
70 |
80 |
11 |
|
5 |
>
49.6 |
49.6 |
38.1 |
>
45.3 |
45.3 |
38.30 |
83 |
76 |
62 |
|
6 |
>
47.1 |
47.1 |
26.2 |
>
45.3 |
45.3 |
38.30 |
83 |
45 |
60 |
|
7 |
22.6 |
46.4 |
62.6 |
>
42.2 |
42.2 |
38.39 |
54 |
94 |
57 |
|
8 |
>
59.1 |
59.1 |
36.1 |
>
54.4 |
54.4 |
22.28 |
60 |
83 |
NT |
|
9 |
>
46.2 |
46.2 |
31.6 |
>
36.3 |
36.2 |
20.36 |
62 |
96 |
49 |
|
10 |
>
69.7 |
69.7 |
26.6 |
>
55.9 |
55.9 |
21.38 |
88 |
84 |
38 |
|
11 |
>
61.6 |
61.6 |
20.1 |
NT |
NT |
NT |
51 |
98 |
69 |
|
12 |
5.6 |
72.6 |
126 |
>
55.1 |
55.1 |
48.48 |
62 |
80 |
6 |
|
13 |
>
96.2 |
96.2 |
39.6 |
>
123 |
123.0 |
22.24 |
59 |
100 |
10 |
|
14 |
12.3 |
64.6 |
68.4 |
NT |
NT |
NT |
88 |
81 |
100 |
|
15 |
7.6 |
92.1 |
93.6 |
>
81.9 |
81.9 |
35.73 |
80 |
80 |
100 |
|
16 |
19.2 |
34.6 |
56.6 |
NT |
NT |
NT |
86 |
80 |
100 |
NT
indicates not tested
a50%
Effective concentration, or concentration required to inhibit HIV-1 induced
cytopathicity cell lines by 50%.
b50%
Cytotoxic
concentration, or concentration required to reduce the viability of
mock-infected cell lines by 50%.
Table
5. In vitro antibacterial activity
(MICs in μM)
|
Microorganism |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
|
K. ozaenae |
39.00 |
42.9 |
18.2 |
17.7 |
0.64 |
17.8 |
0.009 |
0.555 |
4.64 |
1.16 |
|
K. pneumoniae |
39.00 |
42.9 |
18.2 |
17.7 |
5.12 |
17.8 |
0.555 |
17.8 |
18.6 |
18.6 |
|
S. sonnei |
39.00 |
42.9 |
18.2 |
17.7 |
0.64 |
1.11 |
0.278 |
0.278 |
0.0091 |
0.0091 |
|
Plesiomonas |
39.00 |
42.9 |
0.569 |
2.21 |
0.64 |
17.8 |
0.555 |
0.278 |
18.6 |
18.6 |
|
S. boydii |
39.00 |
42.9 |
18.2 |
17.7 |
10.2 |
17.8 |
1.11 |
17.8 |
37.2 |
18.6 |
|
M. morganii |
39.00 |
42.9 |
18.2 |
17.7 |
20.5 |
17.8 |
1.11 |
35.6 |
37.2 |
18.6 |
|
S. aureus |
39.00 |
42.9 |
18.2 |
17.7 |
20.5 |
17.8 |
1.11 |
0.555 |
2.32 |
1.16 |
|
P.
aeroginosa |
39.00 |
42.9 |
36.4 |
17.7 |
20.5 |
35.6 |
0.278 |
35.6 |
37.2 |
74.2 |
|
V. mimicus |
2.44 |
1.34 |
0.569 |
0.0086 |
0.32 |
0.0086 |
0.139 |
0.0086 |
0.0091 |
0.0091 |
|
V. fluvialis |
2.44 |
2.68 |
0.009 |
0.0086 |
0.32 |
0.0086 |
0.0086 |
0.0086 |
0.00907 |
0.0091 |
|
V. cholerae 0139 |
2.44 |
0.021 |
0.009 |
0.0173 |
0.32 |
0.0086 |
0.0086 |
0.0086 |
0.00907 |
0.0091 |
|
V. cholerae 01 |
1.22 |
0.0048 |
2.28 |
1.10 |
1.28 |
2.22 |
0.0086 |
0.278 |
1.16 |
0.145 |
|
V. parahaemolyticus |
2.44 |
2.68 |
1.14 |
2.21 |
2.56 |
1.11 |
0.0086 |
0.0086 |
0.58 |
1.16 |
|
E. Coli NCTC10418 |
39.00 |
42.9 |
18.2 |
17.7 |
20.5 |
17.8 |
0.0086 |
35.6 |
37.2 |
18.6 |
|
E. tarda |
39.00 |
42.9 |
36.4 |
17.7 |
20.5 |
8.89 |
0.139 |
0.139 |
37.2 |
18.6 |
|
P. vulgaris |
39.00 |
42.9 |
18.2 |
17.7 |
20.5 |
8.89 |
0.278 |
2.22 |
37.2 |
18.6 |
|
P. mirabilis |
39.00 |
42.9 |
18.2 |
17.7 |
20.5 |
17.8 |
0.278 |
71.1 |
37.2 |
18.6 |
|
S. typhimurium |
39.00 |
42.9 |
18.2 |
8.84 |
20.5 |
35.6 |
8.89 |
71.1 |
37.2 |
18.6 |
|
S. paratyphi A |
39.00 |
10.7 |
18.2 |
17.7 |
2.56 |
2.22 |
0.139 |
2.22 |
0.29 |
0.58 |
|
S. typhi |
39.00 |
42.9 |
18.2 |
17.7 |
20.5 |
17.8 |
0.009 |
71.1 |
37.2 |
18.6 |
|
S. enteritidis |
19.5 |
42.9 |
18.2 |
17.7 |
20.5 |
0.0086 |
0.139 |
2.22 |
9.29 |
4.64 |
|
C. ferundii |
0.019 |
0.61 |
2.28 |
2.21 |
0.64 |
1.11 |
0.278 |
2.22 |
0.29 |
0.58 |
|
Enterobacter |
0.0381 |
42.9 |
0.569 |
8.84 |
10.2 |
1.11 |
0.0086 |
8.89 |
4.64 |
4.64 |
|
B. megatherius |
19.5 |
42.9 |
18.2 |
17.7 |
10.2 |
0.0086 |
0.0086 |
0.278 |
18.6 |
9.28 |
Table 6.
In vitro antibacterial activity (MIC’s
in μM)
|
Microorganism |
11 |
12 |
13 |
14 |
15 |
16 |
Cipro |
|
Gati |
|
K.
ozaenae |
0.527 |
20.9 |
21.5 |
0.0290 |
0.0002 |
0.0017 |
0.0092 |
0.0629 |
0.0037 |
|
K.
pneumoniae |
16.9 |
20.9 |
21.5 |
0.0002 |
0.0035 |
0.0138 |
0.0023 |
0.1259 |
0.0037 |
|
S.
sonnei |
0.0082 |
20.9 |
21.5 |
0.00003 |
0.0035 |
0.0017 |
0.0023 |
2.0156 |
0.0037 |
|
Plesiomonas |
16.9 |
20.9 |
21.5 |
0.0036 |
0.0002 |
0.0035 |
0.0023 |
0.0629 |
0.1182 |
|
S.
boydii |
33.8 |
20.9 |
43.0 |
0.0002 |
0.0002 |
0.0008 |
0.0023 |
0.5039 |
0.0590 |
|
M.
morganii |
33.8 |
20.9 |
43.0 |
0.0002 |
0.0071 |
0.0008 |
0.0023 |
0.0629 |
0.0009 |
|
S.
aureus |
1.05 |
20.9 |
43.0 |
0.0004 |
0.0071 |
0.0008 |
0.0023 |
0.0314 |
0.0009 |
|
P. aeroginosa |
67.5 |
2.62 |
86.0 |
0.0004 |
0.0071 |
0.0278 |
0.0092 |
0.2519 |
0.0074 |
|
V.
mimicus |
0.0082 |
0.0102 |
0.0105 |
0.0290 |
0.0002 |
0.0017 |
0.0023 |
0.0629 |
0.0074 |
|
V.
fluvialis |
0.0082 |
1.31 |
1.31 |
0.0004 |
0.0566 |
0.0008 |
0.0023 |
0.0629 |
0.0009 |
|
V.
cholerae 0139 |
0.0082 |
0.0102 |
0.0105 |
0.0290 |
0.0002 |
0.0017 |
0.0023 |
0.1259 |
0.0009 |
|
V.
cholerae 01 |
0.0082 |
0.0102 |
0.0105 |
0.0290 |
0.0566 |
0.0069 |
0.0023 |
0.0009 |
0.0009 |
|
V.
parahaemolyticus |
0.527 |
0.327 |
0.672 |
0.0004 |
0.0018 |
0.0035 |
0.0023 |
2.0156 |
0.4727 |
|
E.
Coli NCTC10418 |
33.8 |
20.9 |
86.0 |
0.0004 |
0.0035 |
0.0017 |
0.0011 |
0.0314 |
0.0009 |
|
E.
tarda |
16.9 |
20.9 |
43.0 |
0.0004 |
0.0071 |
0.0035 |
0.0023 |
0.2519 |
0.0009 |
|
P.
vulgaris |
16.9 |
20.9 |
43.0 |
0.0004 |
0.0002 |
0.0008 |
0.0023 |
0.0314 |
0.0009 |
|
P.
mirabilis |
16.9 |
20.9 |
43.0 |
0.0004 |
0.0002 |
0.0035 |
0.0023 |
0.1259 |
0.0009 |
|
S.
typhimurium |
16.9 |
20.9 |
43.0 |
0.0002 |
0.0566 |
0.0035 |
0.0023 |
0.2519 |
0.0009 |
|
S.
paratyphi A |
4.22 |
20.9 |
21.5 |
0.0036 |
0.0142 |
0.0069 |
0.0023 |
0.0314 |
0.0009 |
|
S.
typhi |
16.9 |
20.9 |
21.5 |
0.0002 |
0.00002 |
0.0004 |
0.0023 |
0.5039 |
0.0009 |
|
S.
enteritidis |
8.44 |
0.654 |
21.5 |
0.0002 |
0.00002 |
0.0008 |
0.0023 |
1.0078 |
0.0009 |
|
C.
ferundii |
0.527 |
2.62 |
1.34 |
0.0290 |
0.0071 |
0.0002 |
0.0023 |
1.0078 |
0.0037 |
|
Enterobacter |
0.264 |
20.9 |
21.5 |
0.0002 |
0.0035 |
0.0004 |
0.0023 |
1.0078 |
0.0009 |
|
B.
megatherius |
16.9 |
20.9 |
21.5 |
0.0002 |
0.00002 |
0.0004 |
0.0023 |
1.0078 |
0.0037 |
Table 7. In vitro antibacterial study on E.coli NCTC 10419 strain
|
Compound |
In
Vitro MIC ( in mM / ml) |
In Vivo EC50 (in mg / Kg body wt.) |
|
14 |
0.0004 |
0.46 |
|
15 |
0.0035 |
1.87 |
|
Ciprofloxacin |
0.0011 |
1.25 |
|
Lomefloxacin |
0.0314 |
1.87 |
REFERENCES
[1] Broder S,
Gallo RC: A pathogenic retrovirus (HTLV-III) linked to AIDS. N. Engl. J.
Med. 1984, 311:1292-1297.
[2] Bowen DL,
Hane HC, Fauci AC: Immunopathogenesis of the acquired immunodeficiency syndrome. Ann. Intern.
Med. 1985, 163: 704-709.
[3] Di
Bisceglie A.: Hepatitis C. Lancet 1998, 351: 351-355.
[4] Rosado RR,
Olmeda PM, Samaniego GJ, Soriano V: Management of hepatitis C in HIV-infected persons. Antiviral Res. 2001, 52:
184-198.
[5] Sriram
D, Yogeeswari P: Towards the Design and Development of Agents with Broad
Spectrum Chemotherapeutic Properties for the Effective Treatment of HIV / AIDS. Curr.
Med. Chem. 2003, 10:1909-1915.
[6] De Clercq E:
Perspectives of non-nucleoside reverse transcriptase inhibitors in the therapy
of HIV-1 infection. Farmaco 1999, 54: 26-45.
[8] Tripos
Alchemy 2000, Alchemy 32 Version 2.0, Copyright 1997, Tripos Inc.
[9] Pandeya SN,
Sriram D, De Clercq E, Pannecouque C, Witrouw M: Anti-HIV activity of some Mannich bases
of isatin derivatives.
[10] Pandeya SN,
Yogeeswari P, Sriram D, De Clercq E, Pannecouque C, Witrouw M: Synthesis and
Screening for Anti-HIV activity
of some N-Mannich bases of isatin derivatives. Chemother. 1999, 45: 192-196.
[11] Balzarini J,
Perez PMJ, Felix SA, Camarasa MJ, Batharst IC, Barr PJ: Kinetics of inhibition
of human immunodeficiency virus type 1 (HIV-1) reverse transcriptase by the
novel HIV-1-specific nucleoside analogue [2',5'-bis-O-(tert-butyldimethylsilyl)-beta-D-ribofuranosyl]-3'-spiro-5
"-
(4"-amino-1",2"-oxathiole-2",2"-dioxide)thymine
(TSAO-T). J.
Biol. Chem. 1992, 267: 11831-11838.
[12] Garcia CA,
Micklatcher M, Turpin JA, Stup TL, Watson K, Buckheit RW: Novel Modifications
in the Alkenyldiarylmethane (ADAM) Series of Non-Nucleoside Reverse
Transcriptase Inhibitors. J. Med. Chem. 1999, 42: 4861-4874.
[13] Guo JT,
Bichkow VV, Seeger C: Effect of alpha interferon on the hepatitis
C virus replicon. J. Virol. 2002, 75:
8516-8522.
[14] Sriram
D, Jyothimallika K, Yogeeswari P: Synthesis of Tetrahydro-2H-[1, 3, 5]
thiadiazine-5-(4-pyridylcarboxamido)-2-thione with antitubercular activity. Sci. Pharm. 2004, 72:
35-41.
[15] Pandeya SN,
Sriram D: Synthesis and screening for antibacterial activity of Schiff’s and
Mannich bases of Isatin and its derivatives. Acta Pharm.
Turc. 1998, 40: 33-38.
[16] Pandeya SN, Sriram D, Nath G, De Clercq E: Synthesis, antibacterial, antifungal and anti-HIV activities of norfloxacin Mannich bases. Eur. J. Med. Chem. 2000, 35: 249-265.
Published by the Canadian Society for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences.