J Pharm Pharmaceut Sci (www.cspscanada.org) 9(2):169-189, 2006
In vitro - In vivo Correlation: From Theory to Applications
Jaber Emami
Department of Pharmaceutics, Faculty of Pharmacy and Pharmaceutical Sciences, Isfahan University of Medical Sciences, Isfahan, Iran
Received March 20, 2006; Revised April 28, 2006; Accepted June 13, 2006; published June 16, 2006.
Correspondence:Dr. Jaber Emami Department of Pharmaceutics, Faculty of Pharmacy and Pharmaceutical Sciences, Isfahan University of Medical Sciences, Isfahan, Iran, E-mail: emami@pharm.mui.ac.ir
ABSTRACT: A key goal in pharmaceutical development of dosage forms is a good understanding of the in vitro and in vivo performance of the dosage forms. One of the challenges of biopharmaceutics research is correlating in vitro drug release information of various drug formulations to the in vivo drug profiles (IVIVC). Thus the need for a tool to reliably correlate in vitro and in vivo drug release data has exceedingly increased. Such a tool shortens the drug development period, economizes the resources and leads to improved product quality. Increased activity in developing IVIVCs indicates the value of IVIVCs to the pharmaceutical industry. IVIVC can be used in the development of new pharmaceuticals to reduce the number of human studies during the formulation development as the main objective of an IVIVC is to serve as a surrogate for in vivo bioavailability and to support biowaivers. It supports and/or validates the use of dissolution methods and specification settings. This is because the IVIVC includes in vivo relevance to in vitro dissolution specifications. It can also assist in quality control for certain scale-up and post-approval changes (SUPAC). With the proliferation of modified-release products, it becomes necessary to examine the concept of IVIVC in greater depth. Investigations of IVIVC are increasingly becoming an integral part of extended release drug development. There must be some in vitro means of assuring that each batch of the same product will perform identically in vivo. This review article represents the FDA guidance, development, evaluation, and validation of an IVIVC to grant biowaivers, and to set dissolution specifications for oral dosage forms, biopharmaceutics classification systems (BCS), BCS biowaivers, application of BCS in IVIVC development and concept of mapping. The importance of dissolution media and methodology and pharmacokinetic studies in the context of IVIVC has been highlighted. The review also covers the literature examples of IVIVCs regarding internal and external validation, compendial dissolution assessment, formulation dependency of IVIVCs, and IVIVCs of pure enantiomers versus racemate drugs. The same principles of IVIVC used for oral extended release products may be applied for non-oral products such as parenteral depot formulations and novel drug delivery systems as well.
INTRODUCTION
In recent years, the concept and application of the in vitro-in vivo correlation (IVIVC) for pharmaceutical dosage forms have been a main focus of attention of pharmaceutical industry, academia, and regulatory sectors. Development and optimization of formulation is an integral part of manufacturing and marketing of any therapeutic agent which is indeed a time consuming and costly process. Optimization process may require alteration in formulation composition, manufacturing process, equipment and batch sizes. If these types of changes are applied to a formulation, studies in human healthy volunteers may be required to prove that the new formulation is bioequivalent with the old one. Certainly, implementation of these requirements not only halts the marketing of the new formulation but also increases the cost of the optimization processes. It would be, desirable, therefore, to develop in vitro tests that reflect bioavailability data. A regulatory guidance for both immediate- and modified-release dosage forms has been, therefore, developed by the FDA to minimize the need for bioavailability studies as part of the formulation design and optimization. IVIVC procedures are specific to certain countries but could be adopted or used as the background for regulatory recommendations by other countries. IVIVC can be used in the development of new pharmaceuticals to reduce the number of human studies during the formulation development. The main objective of an IVIVC is to serve as a surrogate for in vivo bioavailability and to support biowaivers. IVIVCs could also be employed to establish dissolution specifications and to support and/or validate the use of dissolution methods.This is because the IVIVC includes in vivo relevance to in vitro dissolution specifications. It can also assist in quality control for certain scale-up and post-approval changes, for instance, to improve formulations or to change production processes. There must be some in vitro means of assuring that each batch of the same product will perform identically in vivo. With the proliferation of modified-release products, it is essential to examine the concept of IVIVC in greater depth. Therefore, a more detailed article covering various aspects of an IVIVC study including complete process of developing the correlation with high quality, accurate and precise predictability, and identifying specific applications for such correlations might be of importance. Although the focus of discussion, in this review, will primarily be centered on modified-release formulations for which IVIVC is believed to be more defined, various aspects of the IVIVC of immediate-release dosage forms are also discussed.
DEFINITIONS
The term correlation is frequently employed within the pharmaceutical and related sciences to describe the relationship that exists between variables. Mathematically, the term correlation means interdependence between quantitative or qualitative data or relationship between measurable variables and ranks (1). From biopharmaceutical standpoint, correlation could be referred to as the relationship between appropriate in vitro release characteristics and in vivo bioavailability parameters. Two definitions of IVIVC have been proposed by the USP and by the FDA (2, 3).
United State Pharmacopoeia (USP) definition
The establishment of a rational relationship between a biological property, or a parameter derived from a biological property produced by a dosage form, and a physicochemical property or characteristic of the same dosage form (2).
Food and Drug Administration (FDA) definition
IVIVC is a predictive mathematical model describing the relationship between an in vitro property of a dosage form and a relevant in vivoresponse. Generally, the in vitro property is the rate or extent of drug dissolution or release while the in vivo response is the plasma drug concentration or amount of drug absorbed (3).
CORRELATION LEVELS
Five correlation levels have been defined in the IVIVC FDA guidance (3). The concept of correlation level is based upon the ability of the correlation to reflect the complete plasma drug level-time profile which will result from administration of the given dosage form (2).
Level A Correlation
This level of correlation is the highest category of correlation and represents a point-to-point relationship between in vitro dissolution rate and in vivo input rate of the drug from the dosage form (2). Generally, percent of drug absorbed may be calculated by means of model dependent techniques such as Wagner-Nelson procedure or Loo-Riegelman method or by model-independent numerical deconvolution (2). These techniques represent a major advance over the single-point approach in that these methodologies utilize all of the dissolution and plasma level data available to develop the correlations (2) and will be discussed more in detail later in this article. The purpose of Level A correlation is to define a direct relationship between in vivo data such that measurement of in vitro dissolution rate alone is sufficient to determine the biopharmaceutical rate of the dosage form. In the case of a level A correlation, an in vitro dissolution curve can serve as a surrogate for in vivo performance. Therefore, a change in manufacturing site, method of manufacture, raw material supplies, minor formulation modification, and even product strength using the same formulation can be justified without the need for additional human studies (2). It is an excellent quality control procedure since it is predictive of the dosage form’s in vivo performance.
Level B Correlation
A level B IVIVC utilizes the principles of statistical moment analysis. In this level of correlation, the mean in vitro dissolution time (MDTvitro) of the product is compared to either mean in vivo residence time (MRT) or the mean in vivo dissolution time (MDTvivo). MRT, MDTvitro and MDTvivo will be defined throughout the manuscript where appropriate. Although a level B correlation uses all of the in vitro and in vivo data, it is not considered to be a point-to-point correlation, since there are a number of different in vivo curves that will produce similar mean residence time values (3). A level B correlation does not uniquely reflect the actual in vivo plasma level curves. Therefore, one can not rely upon a level B correlation alone to justify formulation modification, manufacturing site change, excipient source change, etc. In addition in vitro data from such a correlation could not be used to justify the extremes of quality control standards (2).
Level C Correlation
In this level of correlation, one dissolution time point (t50%, t90%, etc.) is compared to one mean pharmacokinetic parameter such as AUC, tmax or Cmax. Therefore, it represents a single point correlation and doses not reflect the entire shape of the plasma drug concentration curve, which is indeed a crucial factor that is a good indicative of the performance of modified-release products (2, 3). This is the weakest level of correlation as partial relationship between absorption and dissolution is established. Due to its obvious limitations, the usefulness of a Level C correlation is limited in predicting in vivo drug performance. The usefulness of this correlation level is subject to the same caveats as a Level B correlation in its ability to support product and site changes as well as justification of quality control standard extremes (2). Level C correlations can be useful in the early stages of formulation development when pilot formulations are being selected. While the information may be useful in formulation development, waiver of an in vivo bioequivalance study (biowaiver) is generally not possible (3).
Multiple-level C correlation
A multiple level C correlation relates one or several pharmacokinetic parameters of interest (Cmax, AUC, or any other suitable parameters) to the amount of drug dissolved at several time points of the dissolution profile. A multiple point level C correlation may be used to justify a biowaiver, provided that the correlation has been established over the entire dissolution profile with one or more pharmacokinetic parameters of interest. A relationship should be demonstrated at each time point at the same parameter such that the effect on the in vivo performance of any change in dissolution can be assessed (3). If such a multiple level C correlation is achievable, then the development of a level A correlation is also likely. A multiple Level C correlation should be based on at least three dissolution time points covering the early, middle, and late stages of the dissolution profile.
Level D correlation
Level D correlation is a rank order and qualitative analysis and is not considered useful for regulatory purposes. It is not a formal correlation but serves as an aid in the development of a formulation or processing procedure (3, 4).
SYSTEMATIC DEVELOPMENT OF A CORRELATION
Any well designed and scientifically sound approach would be acceptable for establishment of an IVIV correlation (2). As the development of an IVIVC is a dynamic process starting from the very early stages of development program through the final step, the following practical and detailed approach with industrial application is summarized from reference number 5 without modifications.
"To understand how an IVIVR is used throughout the product development cycle, it is useful to become familiar with the following terms as they relate to a typical product development cycle for oral extended-release product (Fig. 1). An assumed IVIVR is essentially one that provides the initial guidance and direction for the early formulation development activity. Thus, during stage 1 and with a particular product concept in mind, appropriate in vitro targets are established to meet the desired in vivo profile specification. This assumed model can be the subject of revision as prototype formulations are developed and characterized in vivo, with the results often leading to a further cycle of prototype formulation and in vivo characterization. Out of this cycle and in vivo characterization and, of course, extensive in vitro testing is often developed what can be referred to as retrospective IVIVR. With a defined formulation that meets the in vivo specification, Stage 2 commences. At this stage based on a greater understanding and appreciation of defined formulation and its characteristics, a prospective IVIVR is established through a well defined prospective IVIVR study. Once the IVIVR is established and defined it can be then used to guide the final cycle of formulation and process optimization leading into Stage 3 activities of scale-up, pivotal batch manufacture, and process validation culminating in registration, approval and subsequent post-approval scale-up and other changes. Thus rather than viewing the IVIVR as a single exercise at a given point in a development program, one should view it as a parallel development in itself starting at the initial assumed level and being built on and modified through experience and leading ultimately to a prospective IVIVR".
"Stage 1: To undertake the development of an oral extended-release product, stage 1 targets first must be defined. The target in vivo profile needs to be first established, based on, if possible, pharmacokinetic/pharmacodynamic models. Clearly, as described in the pioneering work of Amidon in relation to the original biopharmaceutic drug classification and the work of Corrigan relating to extended release product, characterizing the permeability properties of a drug substance is a key element both in establishing the initial feasibility of any formulation program and in the subsequent interpretation of the observed in vivo absorption characteristics of a given dosage form. The physicochemical characteristics of the drug substance itself, in the context of how these affect the formulation approach and in the context of relevance to dissolution at distal sites in the gastro-intestinal tract, need to be taken into account. Based on this information a priori in vitro methods are usually then developed and a theoretical in vitro target is established, which should achieve the desired absorption profile. Essentially at this stage a level A correlation is assumed and the formulation strategy is initiated with the objective of achieving the target in vitro profile. The prototype formulation program itself is normally initiated with some knowledge or expectation of what technologies and/or mechanism of release are particularly suited to meet the desired targets. This work is usually done at a laboratory level of manufacture with the simplest dissolution methodology that seems appropriate. Prototypes that meet the target in vitro profile are then selected involving one or, very often, more than one technology or formulation approach. At least one, but usually more than one prototype within each technology or formulation approach is tested. More extended in vitro characterization, which looks at the robustness of these prototypes across dissolution conditions such as pH, medium, agitation speed and apparatus type, is routine at this point. Certainly, stage 1 activity should culminate in a pilot PK study. This is typically a four or five-arm cross-over study. The size of this pilot pharmacokinetic study will vary depending on the inherent variability of the drug itself but typically range from 6 to 10 subjects. The results of this pilot PK study provide the basis for establishing what has been referred to as a retrospective IVIVR. In other words, a number of different prototypes with some level of variation in release rate have now been characterized both in vitro and in vivo. This information first allows a reality check on both the in vivo and assumed IVIVR, either matching expectation or often causing a fundamental shift in the assumed IVIVR. After the results of the in vivo study are known, there is often a phase of significant revision of the in vitro methods, sometimes driven by the need to detect an in vitro difference that was observed in vivo but that had not been detected using the original in vitro methods. This work sometimes results in revised in vitro targets and reformulation strategy and the same cycle of activity again".
"Stage 2: By this stage of the development process, a defined formulation that meets the in vivo targets has been achieved. The aim is to progress through the normal formulation process optimization steps ultimately into scale-up, registration, and approval. In stage 2, a defined formulation and ideally a good understanding of the mechanism of release of this formulation has been established. Based on this a priori understanding, and from a sort of retrospective data generated from stage 1, an empirical basis exist for determining the primary formulation related rate controlling variables. For extended-release products, this a priori understanding is usually more obvious than might be the case for immediate-release products. Based on this information, a number of products with different release rates are usually manufactured by varying the primary rate controlling variable but within the same qualitative formulation. Extensive in vitro characterization is again performed across pH, media and apparatus, but the stage 1 work is also taken into account. This leads to execution of a prospective IVIVR study. The IVIVR is developed and defined after an analysis of the result of that prospective in vivo study. It can often involved further in vitro method development in the context of the observed results, but clearly with the objective of establishing a definitive IVIVR. This ideally is a level A IVIVC but, in particular, multiple-level C IVIVC continues to be both an acceptable and useful IVIVR. This work should also result in the definitive in vitro method that has been shown to be correlated with in vivo performance and sensitive to the specific formulation variables.

Fig. 1: The product development process for extended-release products (from reference 5 with permission).
Once the IVIVR is established, it is routinely used in the completion of the formulation/process optimization program using statistically based experimental design studies looking at critical formulation and process variables and their interactions. By now with a correlated in vitro method, the robustness of the formulation and process can be established. This information can also be used to establish appropriate in-process and finished-product specification, of course, the appropriate targets for scale-up". Development of in vitro in vivo correlation and validation using in vitro dissolution and in vivo time course is also illustrated in Figure 2.
IMPORTANT CONSIDERATIONS IN DEVELOPING A CORRELATION
When the dissolution is not influenced by factors such as pH, surfactants, osmotic pressure, mixing intensity, enzyme, ionic strength, a set of dissolution data obtained from one formulation is correlated with a deconvoluted plasma concentration-time data set (3). To demonstrate a correlation, fraction absorbed in vivo should be plotted against the fraction released in vitro. If this relationship becomes linear with a slope of 1, then curves are superimposable, and there is a 1:1 relationship which is defined as point-to-point or level A correlation. Under these circumstances, the correlation is considered general and could be extrapolated within a reasonable range for that formulation of the active drug entity.
In a linear correlation, the in vitro dissolution and in vivo input curves may be directly superimposable or may be made to be superimposable by the use of appropriate scaling factor (time corrections) (2, 3). Time scaling factor should be the same for all formulations and different time scales for each formulation indicate absence of an IVIVC (3). Non-linear correlation may also be appropriate (2, 3).
In cases where, the dissolution rate depends on the experimental factors mentioned above, the deconvoluted plasma concentration-time curves constructed following administration of batches of product with different dissolution rates (at least two formulations having significantly different behavior) are correlated with dissolution data obtained under the same dissolution condition. If there is no one-to-one correlation other levels of correlation could be evaluated (2, 3).
If one or more of the formulations (highest or lowest release rate formulations) may not illustrate the same relationship between in vitro performance and in vivo profiles compared with the other formulations, the correlation is still valid within the range of release rates covered by the remaining formulations (3).
The in vitro dissolution methodology should be able to adequately discriminate between the study formulations. Once a system with most suitable discrimination is developed, dissolution conditions should be the same for all formulations tested in the biostudy for development of the correlation (3).
During the early stages of correlation development, dissolution conditions may be altered to attempt to develop a one-to-one correlation between the in vitro dissolution profile and the in vivo dissolution profile (3).
An established correlation is valid only for a specific type of pharmaceutical dosage form (tablets, gelatin capsules, etc.) with a particular release mechanism (matrix, osmotic system, etc.) and particular main excipients and additives. The correlation is true and predictive only if modifications of this dosage form remain within certain limits, consistent with the release mechanism and excipients involved in it (3).
Extrapolation of IVIVC established in healthy subjects to patients has to be taken into account. Drugs are often taken just before, with or after meal. All these factors may increase variability. A posterior correlation might be established using the patients' data only to increase the knowledge of the drug.
The release rates, as measured by percent dissolved, for each formulation studied, should differ adequately (e.g., by 10%). This should result in vivo profiles that show a comparable difference, for example, a 10% difference in the pharmacokinetic parameters of interest (Cmax or AUC) between each formulation (3).
BIOPHARMACEUTICS CLASSIFICATION SYSTEM (BCS)
The Biopharmaceutics Classification System (BCS) is a drug development tool that allows estimation of the contribution of three fundamental factors including dissolution, solubility and intestinal permeability, which govern the rate and extent of drug absorption from solid oral dosage forms (6). Drug dissolution is the process by which the drug is released, dissolved and becomes ready for absorption. Permeability is referred to the ability of the drug molecule to permeate through a membrane in to the systemic circulation. BCS is also a fundamental guideline for determining the conditions under which IVIVCs are expected. It is also used as a tool for developing the in-vitrodissolution specification (6, 7). The classification is dealing with drug dissolution and absorption model, which considers the key parameters controlling drug dissolution and absorption as a set of dimensionless numbers: the absorption number, the dissolution number, and the dose number (6, 7).
Absorption Number (An)
The Absorption Number (An) is the ratio of the Mean Residence Time (Tres) to the Mean Absorption Time (Tabs) and is calculated by equation 1
![]() (1)
(1)
Example: Calculate An, considering following parameters: Peff = 1 × 10-3 cm/sec, Tres = 180 min, and R = 1 cm, therefore, An = 180/ (1/10-3) = 10.
Table 1: Calculated parameters for representative drugs. From Reference 6 with permission. |
|||||
Drug |
Dose (mg) |
Csmin (mg/ml) a |
Vsol |
Doc |
Dnd |
Piroxicam |
20 |
0.007 |
2857 |
11.4 |
0.15 |
Glyburide |
20 |
<0.10 |
133 |
>0.8 |
0.78 |
Cimetidine |
800 |
6.000 |
556 |
0.53 |
129 |
Chlorthiazide |
500 |
0.786 |
636 |
2.54 |
17.0 |
Digoxin |
0.5 |
0.024 |
20.8 |
0.08 |
0.52 |
Griseofulvin |
500 |
0.015 |
33333 |
133 |
0.32 |
Carbamazepine |
200 |
0.260 |
769 |
3.08 |
5.61
|
a Minimum physiologic solubility determined in physiological pH range of 1-8 and temperature 37°C (8, 9); b Volume of solvent required to completely dissolve the dose at minimum physiologic solubility; c Vo = 250 ml; d Assumptions: ro = 25 μm, D = 5*10-6 cm2/sec, ρ = 1.2 gm/cm3, Tres = 180 min (10). Numbers in parentheses are references. reference |
|||||
Dissolution Number (Dn)
The Dissolution Number (Dn) is the ratio of Tres to Mean Dissolution Time (Tdiss) and could be estimated using equation 2.
![]() (2)
(2)
Example: Refer to table 1.
Dose Number (Do)
The Dose Number (Do) is calculated using equation 3:
![]() (3)
(3)
Example: Refer to table 1.
Where: L = tube length, R = tube radius, π = 3.14, Q = fluid flow rate, ro = initial particle radius, D = particle acceleration, ρ = particle density, Peff = effective permeability, Vo is the initial gastric volume equal to 250 ml which is derived from typical bioequivalance study protocols that prescribe administration of a drug product to fasting human volunteers with a glass of water at the time of drug administration and Csmin is minimum aqueous solubility in the physiological pH range of 1-8 (6).
The dose, dose number, solubility, and estimated dissolution number for a number of drugs are listed in Table 1. The fraction dose absorbed could be estimated using these three major dimensionless parameters. However, the extent of solubilization and potential particle aggregation in the small intestine is unknown and therefore, the solubility, dose and dissolution number of a drug in vivo is difficult to estimate precisely (6). As the drug dissolution and intestinal permeability are the fundamental parameters governing rate and extent of drug absorption, drugs could be categorized into high/low solubility and permeability classes. Thus, the expectations regarding IVIVC could be stated more clearly as are summarized in Table 2.
Table 2: IVIVC expectations for immediate release products based on BCS (from ref. 6 with modification) |
|||||
Class |
Solubility |
Permeability |
Absorption rate control |
IVIVC expectations for Immediate release product |
|
I |
High |
High |
Gastric emptying |
IVIVC expected, if dissolution rate is slower than gastric emptying rate, otherwise limited or no correlations |
|
II |
Low |
High |
Dissolution |
IVIVC expected, if in vitro dissolution rate is similar to in vivo dissolution rate, unless dose is very high. |
|
III |
High |
Low |
Permeability |
Absorption (permeability) is rate determining and limited or no IVIVC with dissolution. |
|
IV |
Low |
Low |
Case by case |
Limited or no IVIVC is expected. |
|
Class I compounds such as metoprolol exhibit a high absorption (An) and a high Dissolution (Dn) number. The rate-limiting step to drug absorption is drug dissolution or gastric emptying rate if dissolution is very rapid (6). This group of drugs is expected to be well absorbed unless they are unstable, form insoluble complexes, are secreted directly from gut wall, or undergo first pass metabolism (7). For immediate release products that release their content very rapidly the absorption rate will be controlled by the gastric emptying rate and no correlation of in vivo data with dissolution rate is expected (6). Dissolution test for immediate release formulations of class I drugs, therefore, need only to verify that the drug indeed is rapidly released from the dosage form under mild aqueous conditions (7). A dissolution specification of 85% dissolution of drug contained in immediate release in 15 minutes may insure bioequivalance (11-13). The FIP consider a formulation as a very fast releasing when at least 80% of the drug substance is dissolved in a bout 20-30 minutes under reasonable and justified test conditions (13). The aforementioned dissolution time limits are based on typical gastric emptying times for water in the fasted state.
When a class I drug is formulated as an extended release product in which the release profile controls the rate of absorption, and the solubility and permeability of the drug is site independent, a level A correlation is most likely. However, once the permeability is site dependent a level C correlation is expected (14).
Class II drugs such as phenytoin has a high absorption number, An, but a low dissolution number, Dn. In vivodrug dissolution for Class II drugs is, therefore, a rate-limiting factor in drug absorption (except at very high dose number, Do) and consequently absorption is usually slower than Class I and takes place over a longer period of time (6). The limitation can be equilibrium or kinetic in nature. In the case of an equilibrium problem enough fluid is not available in the GI tract to dissolve the dose. For example, 33.3 liters (Table 1) of fluid are required to dissolve one dose of griseofulvin (6, 15). As the total volume of fluid entering the GI tract within 24 hrs period is only about 5 to 10 liters (16), insufficient fluid would be available at any given time to dissolve the entire dose of griseofulvin (7). Griseofulvin exhibits a high dosing number (Do) and a low dissolution number (Dn). Bioavailability and the fraction of the dose absorbed can be improved by decreasing Do by reducing the dose, by taking more water with the administered dose or by increasing drug solubility. The dose of a drug is determined on the basis of pharmacokinetic / pharmacodynamic considerations and could not be altered. The volume of water initially is taken with dosage form will be limited by patient compliance and anatomical and physiological capacity of the stomach. For griseofulvin, therefore, only enhancement of the drug solubility through appropriate formulation approach (i.e. solid dispersion) can lead to reduced Dn considerably and to increase drug bioavailability (17). In the case of kinetic problem, the entire dose of the drug dissolves too slowly. For example a typical dose of digoxin is 0.5 mg and has a Vsol of 20.8 ml which results in a small Do. In spite of the small volume of fluid required to dissolve 0.5 mg of the drug, it is shown that bioavailability of digoxin depends on the particle size. Digoxin exhibits dissolution rate limited absorption (Dn = 0.52) at particle sizes of greater than 10 μ in diameter (7, 15, 18). These comply with the reports indicating that digoxin, in micronized form, and griseofulvin, in ultramicronized form, was almost completely absorbed (18-20). For class II drugs, therefore, a strong correlation between dissolution rate and the in vivo performance could be established (7). As pointed out earlier, the appropriate design of in vitro dissolution tests such that discriminate between formulations with different bioavailabilities plays a major role in the ability of the IVIVC predictability. Therefore, it is essential that in vitro dissolution tests reflect in vivo situations when it is used to establish an IVIVC. Dissolution media and methods that reflect the in vivo controlling process are particularly important in this case if good IVIV correlations are to be obtained. The dissolution profile for class II drugs requires multiple sampling times and the use of more than one dissolution medium. Addition of surfactant to simulate in vivo environment might be required. When a class II drug is formulated as an extended release product and the solubility and permeability of the drug is site independent, a level A correlation is expected (14). However, once the permeability is site dependent little or no IVIV correlation is expected (14).
BCS classifications in conjunction with the numerous of compendial and physiological media available could be employed as a fundamental guidances for designing appropriate biorelevant dissolution conditions leading to a more meaningful prediction of in vivo performances. For class I drugs, simple and mild aqueous dissolution media such as SGF without pepsin is suitable, while milk as dissolution medium might be appropriate for specific food/formulation interaction (21). For neutral class II drugs, the fluid simulating conditions in the proximal intestine in the fasted state (FaSSIF) reflects the dissolution in the upper GI tract under fasted state conditions (21). If a class II drug is a weak base, SGFsp could be used to assess the dissolution of the drug in the stomach under fasted state conditions (21). To verify the possibility of drug precipitation under intestinal conditions, performing dissolution in fasted state intestinal conditions (FaSSIF) may be appropriate (21). Comparison of dissolution results obtained under fasted conditions to those of FeSSIF could be a good indicative of whether the formulation should be administered before or after meals (21). In the case of class II weak acids, dissolution could be performed in FaSSIF as a suitable representative of intestinal fasted state conditions. Milk with its composition of lipids and proteins or FeSSIF containing high bile salt/lecithin levels can be employed to simulate the fed state conditions (21).
Class III drugs, such as cimetidine, are rapidly dissolving and permeability is the rate-controlling step in drug absorption. Rapid dissolution is particularly desirable in order to maximize the contact time between the dissolved drug and absorption mucosa. Therefore, the duration of dissolution should be at least as stringent for class III drugs as for class I drugs (7). Furthermore, Class III drugs exhibit a high variability in rate and extent of absorption, but if dissolution is fast such that 85% of drug dissolves in 15 minutes, the variation could be attributed to GI transit, luminal contents, and membrane permeation rather than dosage form factors (6). As drug permeation is rate controlling, limited or no IVIV correlation is expected.
Class IV drugs are low solubility and low permeability drugs. This class of drugs exhibit significant problems for effective oral delivery. It is anticipated that inappropriate formulation of drugs fall in class IV, as in the case of class II drugs, could have an additional negative influence on both the rate and extent of drug absorption. Thus for all categories, it is anticipated that well-designed dissolution tests can be a key prognostic tool in the assessment of both the drugs potential for oral absorption and of the bioequivalance of its formulations (7).
DISSOLUTION MEDIA AND METHODOLOGY
Drug absorption from a solid dosage form following oral administration depends on the release of the drug substance from the drug product, the dissolution or solubilization of the drug under physiological conditions, and the permeability across the gastrointestinal tract. Because of the critical nature of the first two of these steps, in vitro dissolution may be relevant to the prediction of in vivo performance. The solubility of a drug is determined by dissolving the highest unit dose of the drug in 250 ml of buffer adjusted between pH 1 and 8. A drug substance is considered highly soluble when the dose/solubility volume of solution are less than or equal to 250 ml. In addition, if the extent of drug absorption is greater than 90% given that the drug is stable in the gastrointestinal environment; it will be considered as a high permeable drug (22, 23). With perhaps only few exceptions sink conditions are required to obtain in vitro dissolution curves representing the biopharmaceutical properties of the drug product under investigation with minimal effects due to the influence of solubility.
The purpose of in vitro dissolution studies in drug development process is to assess the lot-to-lot quality of a drug product, guide development of new formulations; and ensure continuing product quality and performance after certain changes, such as changes in the formulation, the manufacturing process, the site of manufacture, and the scale-up of the manufacturing process (23). However, from the IVIVC standpoint, dissolution serves as a surrogate for drug bioavailability. Thus more rigorous dissolution standards may be necessary for the in vivo waiver (22). Generally, a dissolution methodology, which is able to discriminate between the study formulations with different release patterns and best, reflects the in vivo behavior should be used to establish an IVIVC. The in vitro dissolution release of a formulation can be modified to facilitate the correlation development. Changing dissolution testing conditions such as the stirring speed, choice of apparatus, pH of the medium, and temperature may alter the dissolution profile. Once a discriminating system is developed, dissolution conditions should be the same for all formulations tested in the biostudy for development of the correlation and should be fixed before further steps towards correlation evaluation are undertaken (3).
Four basic types of dissolution apparatus including rotating basket (Apparatus 1), paddle method (Apparatus 2), reciprocating cylinder (Apparatus 3) and flow through cell (Apparatus 4) are specified by the USP (2) and recommended in the FDA guidance (23, 24). However the first two methods are preferred and it is recommended to start with the basket or paddle method prior to using the others unless shown unsatisfactory (22, 23). Reciprocating cylinder has been found to be especially for bead type modified-release dosage forms. Apparatus 4 may offer advantages for modified release dosage forms that contain active ingredients with very limited solubility. Apparatus 5 (paddle over disk) and apparatus 6 (cylinder) have been shown to be useful for evaluating and testing transdermal dosage forms (2).
In general an aqueous test medium is preferred (2, 3, 13). The pH of dissolution medium, however, differs slightly between various guidance (2, 3, 13). Water which is allowed by some guidances (2,3,13) or buffered solution preferably not exceeding pH 6.8 is recommended by FDA as the initial medium for development of an IVIVC (3, 23). As recommended by USP, dearated water, a buffered solution (typically pH 4 to 8) or a dilute acid (0.001 to 0.1 N) may preferably be used as dissolution medium for modified-release dosage forms (2). To simulate intestinal fluid or gastric fluid a dissolution medium of pH 6.8 or pH 1.2 should be employed respectively (20). Since the drug solubility depends on the composition of the dissolution medium, surfactants, pH, and buffer capacity play a major role in drug solubility in the GI tract (22). For poorly soluble drugs, therefore, addition of surfactant (e.g., 1% sodium lauryl sulfate) may be appropriate (3, 24, 25). In general, non-aqueous and hydro-alcoholic systems are discouraged unless supported by a documented IVIVC (2, 3, 24-26). More extreme testing conditions (e.g. pH>8) should be justified (3, 13, 23). Strict simulation of physiologic gastrointestinal environment is not recommended and addition of enzyme, salts and surfactants need to be justified (13, 23).
For the IVIVC purposes, the dissolution profiles of at least 12 individual dosage units from each lot should be determined. A suitable distribution of sampling points should be selected to define adequately the profiles. The coefficient of variation (CV) for mean dissolution profiles of a single batch should be less than 10% (3). Since dissolution apparatuses tend to become less discriminative when operated at faster speeds, lower stirring speeds should be evaluated and an appropriate speed chosen in accordance with the test data. Using the basket method the common agitation is 50-100 rpm; with the paddle method, it is 50-75 rpm and 25 rpm for suspension (2, 3, 27).
Comparison between dissolution profiles could be achieved using a difference factor (f1) and a similarity factor (f2) which originates from simple model independent approach (23, 28, 29). The difference factor calculates the percent difference between the two curves at each time point and is a measurement of the relative error between the two curves:
![]() (4)
(4)
Where, n is the number of time points, Rt is the dissolution value of the reference batch at time t, and Tt is the dissolution value of the test batch at time t.
The similarity factor is a logarithmic reciprocal square root transformation of the sum squared error and is a measurement of the similarity in the percent dissolution between the two curves.
![]() (5)
(5)
Generally, f1 values up to 15 (0-15) and f2 values greater than 50 (50-100) ensure sameness or equivalence of the two curves (23).
The mean in vitro dissolution time (MDTvitro) is the mean time for the drug to dissolve under in vitro dissolution conditions. This is calculated using the following equation (3):
![]() (6)
(6)
BIOAVAILABILITY STUDIES FOR DEVELOPMENT OF IVIVC
A bioavailability study should be performed to characterize the plasma concentration versus time profile for each of the formulation (30). Bioavailability studies for IVIVC development should be performed with sufficient number of subjects to characterize adequately the performance of the drug product under study. In prior acceptable data sets, the number of subjects has ranged from 6 to 36. Although crossover studies are preferred, parallel studies or cross-study analyses may be acceptable. The latter may involve normalization with a common reference treatment. The reference product in developing an IVIVC may be an intravenous solution, an aqueous oral solution, or an immediate release product. IVIVCs are usually developed in the fasted state. When a drug is not tolerated in the fasted state, studies may be conducted in the fed state (3). Drug absorption from GI tract following ingestion of an oral dosage form could be influenced by a number of in vivo variables. For the determination of reproducible in vivo parameters and consequently useful in vitro in vivo relationship, it is imperative that such variables be identified. As a result, the study should be designed appropriately that as many variables as possible be eliminated or controlled to prevent or minimize their disturbance of IVIVC. Control or standardization of a number of variables including subject selection criteria such as age, gender, physical condition, etc., and the abstinence by the subject from coffee and other xanthenes containing beverages or food, alcohol, irregular diets and smoking before and during the study should be taken in to consideration. Food, posture and exercise may influence hepatic blood flow which in turn may substantially affect the absorption of drugs possessing high hepatic extraction ratio (31).
As pointed out earlier, one method to develop level A correlation is to estimate the in vivo absorption or dissolution time course using an appropriate deconvolution techniques such as Wagner-Nelson procedure or Loo-Riegelman method or numerical deconvolution for each formulation and subject. Wagner-Nelson and Loo-Riegelman methods are both model dependent in which the former is used for a one-compartment model and the latter is for multi-compartment system. The Wagner-Nelson is less complicated than the Loo-Riegelman as there is no requirement for intravenous data (32). However, misinterpretation on the terminal phase of the plasma profile may be possible in the occurrence of a flip-flop phenomenon in which the rate of absorption is slower than the rate of elimination. According to Wagner-Nelson method, the cumulative fraction of drug absorbed at time t is calculated from Equation 7 as follows:
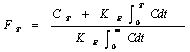 (7)
(7)
Where, CT is plasma concentration at time T and KE is elimination rate constant. The apparent absorption rate constant (Ka) could be obtained from the least square fitted log-linear plot of the percent unabsorbed versus time. The absorption half-life (t1/2a) is calculated as 0.693 / Ka (32, 33).
The Loo-Riegelman method requires drug concentration time data after both oral and intravenous administration of the drug to the same subject and the fraction absorbed at any time t is given by:
![]() (8)
(8)
Where, in addition to symbols defined previously, (Xp)T is the amount of drug in the peripheral compartment as a function of time after oral administration and Vc is the apparent volume of the central compartment. K10, the apparent first order elimination rate constant of drug from the central compartment, is estimated from a previous or subsequent intravenous study of the same subject. (Xp)T/Vc can be estimated by a rather complicated approximation procedure requiring both oral and intravenous data (32).
Deconvolution is a numerical method used to estimate the time course of drug input using a mathematical model based on the convolution integral. For example the absorption rate time course (rabs) that results in plasma concentration (ct) may be estimated by solving the convolution integral equation for rabs.
![]() (9)
(9)
Where, cδ represents the concentration time profile resulting from an instantaneous absorption of a unit amount of drug which is typically from bolus intravenous injection or reference oral solution data, c(t) is the plasma concentration versus time profiles of the tested formulations, rabs is the input rate of the oral solid dosage form in to the body and u is the variable of integration (32, 34, 35). Deconvolution method requires no assumptions regarding of the number of compartments in the model or the kinetics of absorption. Linear distribution and elimination are assumed. Like the Loo-Riegelman method, deconvolution requires data obtained after both oral and intravenous administration in the same subject and assumes no differences in the pharmacokinetics of drug distribution and elimination from one study to the other. Drug concentrations must be measured at the same times following both oral and intravenous administration during the time that drug is absorbed after oral administration (32).
Mean residence time is the mean time that the drug resides in the body and is calculated by following equation:
![]() (10)
(10)
Mean in vivo dissolution time reflects the mean time for drug to dissolve in vivo from a solid dosage form and is estimated as:
![]() (11)
(11)
EVALUATION OF PREDICTABILITY OF IVIVC
An IVIVC should be evaluated to demonstrate that predictability of in vivo performance of a drug product from its in vitro dissolution characteristics is maintained over a range of in vitro dissolution release rates and manufacturing changes (Fig. 2). Since the objective of developing an IVIVC is to establish a predictive mathematical model describing the relationship between an in vitro property and a relevant in vivo response, the proposed evaluation approaches focus on the estimation of predictive performance or, conversely, prediction error. Methodology for the evaluation of IVIVC predictability is an active area of investigation and a variety of methods are possible and potentially acceptable. A correlation should predict in vivo performance accurately and consistently (3).
Depending on the intended application of an IVIVC and the therapeutic index of the drug, evaluation of prediction error internally and/or externally may be appropriate. Evaluation of internal predictability is based on the initial data used to define the IVIVC model (Fig. 2). Evaluation of external predictability is based on additional test data sets (3).
Internal predictability is applied to IVIVC established using formulations with three or more release rates for non-narrow therapeutic index drugs exhibiting conclusive prediction error. If two formulations with different release rates are used to develop IVIVC, then the application of IVIVC would be limited to specified categories (see ref. 3 for categories). Under these circumstances, for complete evaluation and subsequent full application of the IVIVC, prediction of error externally is recommended (3).
External predictability evaluation is not necessary unless the drug is a narrow therapeutic index, or only two release rates were used to develop the IVIVC, or, if the internal predictability criteria are not met i.e. prediction error internally is inconclusive (3,30). However, since the IVIVC will potentially be used to predict the in vivo performance for future changes, it is of value to evaluate external predictability when additional data are available (30).
The objective of IVIVC evaluation is to estimate the magnitude of the error in predicting the in vivo bioavailability results from in vitro dissolution data (Fig. 2). This objective should guide the choice and interpretation of evaluation methods. Any appropriate approach related to this objective may be used for evaluation of predictability (3).
Internal predictability
All IVIVCs should be studied regarding internal predictability. One recommended approach involves the use of the IVIVC model to predict each formulation’s plasma concentration profile (or Cmax and/or AUC for a multiple Level C IVIVC) from each respective formulation’s dissolution data. This is performed for each formulation used to develop the IVIVC model (Fig. 2). Practically, in vitro dissolution rates is first estimated from dissolution data and is converted to in vivo dissolution rates by using the IVIVC model generated slope and intercept. If the cumulative drug release profile is sigmoid, then the Hill equation could be used to parameterize the in vitro drug release.
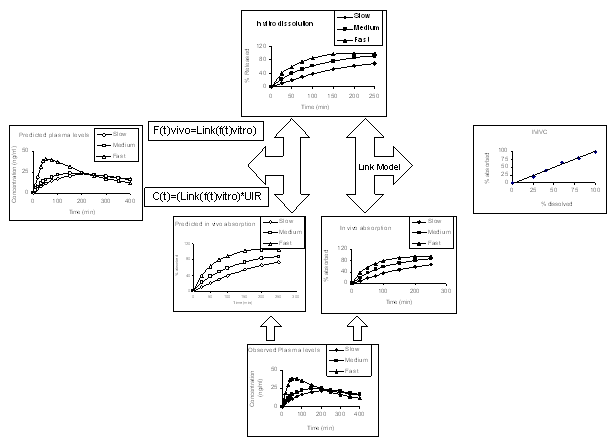
Fig 2: Representative observed and predicted dissolution and plasma profiles which routinely used to develop and validate an in vitro in vivo correlation (based on figures from http://www.uv.es/~mbermejo/DissolutionC with permission).
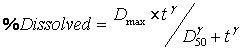 (12)
(12)
Where, %D = the percent drug dissolved at time t, Dmax = the maximum % drug dissolved, D50 = the time required for 50% of the drug to dissolve, t = time and γ = the sigmoidicity factor. In vitro release rates can be calculated by taking the first derivative of the Hill equation as listed below:
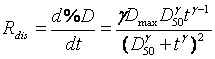 (13)
(13)
The prediction of the plasma concentrations from the corresponding in vivo dissolution profiles is then accomplished by convolution of the in vivo dissolution rates and the pharmacokinetic model for the so called unit impulse response result from i.v. bolus data, oral solution or rapidly releasing (in vivo) immediate release dosage forms using equation 9. In this equation symbols are as previously mentioned. The model predicted bioavailability is then compared to the observed bioavailability for each formulation. The percent prediction errors for Ct, Cmax or AUC could be determined as follows (3, 36-40):
![]() (14)
(14)
Average absolute percent prediction error (% PE) of 10% or less for Cmax and AUC establishes the predictability of the IVIVC. In addition, the % PE for each formulation should not exceed 15%. If these criteria are not met, that is, if the internal predictability of the IVIVC is inconclusive, evaluation of external predictability of the IVIVC should be performed as a final determination of the ability of the IVIVC to be used as a surrogate for bioequivalence (3).
External predictability
Most important when using an IVIVC as a surrogate for bioequivalence is confidence that the IVIVC can predict in vivo performance of subsequent lots of the drug product. Therefore, it may be important to establish the external predictability of the IVIVC. This involves using the IVIVC to predict the in vivo performance for a formulation with known bioavailability that was not used in developing the IVIVC model. % PE of 10% or less for C and AUC establishes the external predictability of an IVIVC. % PE between 10 - 20% indicates inconclusive predictability and the need for further study using additional data sets. Results of estimation of PE from all such data sets should be evaluated for consistency of predictability. % PE greater than 20% generally indicates inadequate predictability, unless otherwise justified (3).
APPLICATION OF AN IVIVC
Biowaivers
The FDA guidance (3) outlines five categories of biowaivers: 1) biowaivers without an IVIVC, 2) biowaivers using an IVIVC: non-narrow therapeutic index drugs, 3) biowaivers using an IVIVC: narrow therapeutic index drugs, 4) biowaivers when in vitro dissolution is independent of dissolution test conditions and 5) situations for which an IVIVC is not recommended for biowaivers (3).
Biowaivers may be granted for manufacturing site changes, equipment changes, manufacturing process changes, and formulation composition changes according to a predictive and reliable IVIVC. The changes may range from minor changes that are not significant to alter product performance to major ones where an IVIVC is not sufficient to justify the change for regulatory decision (30).
Establishment of dissolution specifications
It is relatively easy to establish a multipoint dissolution specification for modified-release dosage forms (2). The dissolution behavior of the biobatch maybe used to define the amount to be released at each time point. However, the difficulty arises in the variation to be allowed around each time point (2). The FDA guidance describes the procedures of setting dissolution specifications in cases of level A, multiple level C, and level C correlation and where there is no IVIV correlation (3).
Once an IVIVC developed, IVIVC should be used to set specifications in such a way that the fastest and lowest release rates allowed by the upper and lower dissolution specifications result in a maximum difference of 20% in the predicted Cmax and AUC. Predicted plasma concentration and consequent AUC and Cmax could be calculated using convolution or any other appropriate modeling techniques (3, 30). In the case of multiple level C correlation, the last time point should be the time point where at least 80% of drug has dissolved (3). For level C correlation, reasonable deviations from ±10 % may be acceptable if the range at any time point does not exceed 25% (3).
When there is no IVIVC, the tolerance limits may be derived from the spread of in vitro dissolution data of batches with demonstrated acceptable in vivo performance (biobatch) or by demonstrating bioequivalance between batches at the proposed upper and lower limit of the dissolution range (the so called side batch concept). Variability in release at each time point is recommended not to exceed a total numerical difference of ±10% (a total of 20%) or less of the labeled claim. In certain cases, deviations from this criterion can be acceptable up to a maximum range of 25%. Beyond this range, the specification should be supported by bioequivalance studies (3, 30).
Concept of mapping
Mapping is a process which relates Critical Manufacturing Variables (CMV), including formulation, processes, and equipment variables that can significantly affect drug release from the product, to a response surface derived from an in vitro dissolution curve and an in vivo bioavailability data (41-43). The mapping process defines boundaries of in vitro dissolution profiles on the basis of acceptable bioequivalency criteria. The goal is to develop product specifications that will ensure bioequivalance of future batches prepared within the limits of acceptable dissolution specifications (23). Dissolution specifications based on mapping would increase the credibility of dissolution as a bioequivalency surrogate marker and will provide continuous assurance and predictability of the product performance. Figure 3 shows the mapping provides for the employment of a dissolution method correlated to the rate and extent of drug bioavailability, which has also been optimized to be sensitive to CMV.
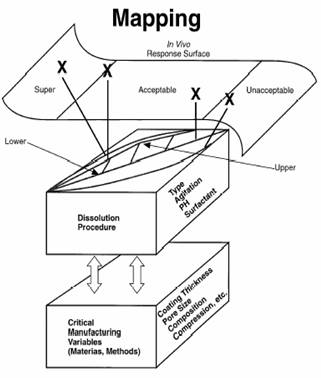
Fig. 3: Mapping, in vitro- in vivo response surface (from reference 40 with permission).
PREVIOUS IVIVC STUDIES
Over four decades ago Levy (44) reported a significant correlation between in vitro dissolution and in vivo bioavailability of aspirin tablets. On the separate study, Wood and others (45) suggested that the drug absorption was very much dependent on dissolution rate. In 1972, Wagner et al demonstrated relationships between in vitro and in vivo pattern of various digoxin dosage forms (46) which was confirmed by other reports (47, 48).
Since then many attempts have been carried out to study the in vitro in vivo correlation for various drugs and dosage forms. The studies have been conducted both in animal, such as rat, rabbit, dog and human. In these studies the possibility of developing different levels of correlation between in vitro dissolution parameters and in vivo pharmacokinetic parameters has been investigated. Although there are many published examples of drugs with dissolution data that correlate well with drug absorption in the body (level A) (33, 36, 37, 49-64), there are also many examples indicating poor correlation of dissolution to drug absorption (level B, 65-66, and level C, 67-77). Multiple level C which is a good acceptable correlation and comparable with level A has also been observed (78, 79). Although numerous level A correlation studies have been reported, most of them suffer from lacking of assessing the predictability of the correlation. Limited number of validated correlation has been reported (36, 37, 52, 61). According to FDA-IVIVC guidance (3), all IVIVCs should be studied in terms of predictability and an average percent prediction error of less than 10% for bioavailability parameters indicates that the correlation is predictive and reliable.
As pointed out the current FDA guideline does not recommend the use of an IVIVC with acceptable internal predictability when manufacturing changes made to a formulation leading to alteration in drug release mechanism. This recommendation is further supported by evaluation of the ability of the IVIVC established for hydrophilic matrix formulations of metoprolol tartrate to predict in vivo performance of a coated bead formulation of the drug with different drug release mechanism (36). The IVIVC model predicted Cmax was 23% higher than observed value which indicates lack of predictability and supports the contention that the IVIVCs are formulation specific.
IVIVC could be employed to examine the appropriateness of the compendial dissolution specifications and the effect of the alteration in dissolution media. A good multiple level C IVIV correlation for L-thyroxine tablet formulation using dissolution method specified in USP 24 has been reported. Alteration in dissolution medium, as proposed in the first supplement of USP 24 and since then (USP25 and USP26), lead to a worse IVIV correlation. It was, therefore, concluded that the old dissolution medium was more discriminative than the newly proposed one (78).
The IVIVC approach has also been utilized in transdermal delivery research to correlate in vitro skin permeation data to the in vivo drug profiles. Systemic drug concentration of 2,3,5,6-tetramethylpyrazine (TMP) following transdermal application in rabbit was successfully accomplished from the in vitro skin permeation data using convolution technique (63). The result of this study demonstrated that the predicted concentrations were in good agreement with the observed drug absorption profile. The findings indicate that in vivo drug profiles could be predicted from the skin permeation tests following transdermal application of the drug.
Various apparatus and methods have been developed to establish IVIVC for biodegradable parenteral delivery systems (80, 81). However, only few examples can be found in the literature where an in vitro drug method accurately predicts the in vivo release profile for parenteral biodegradable depot systems (64, 82). This demonstrates the difficulties in establishing IVIVC for this class of formulations due to the large number of parameters potentially affecting drug release in vivo and in vitro. As, in most cases, diffusion, dissolution and erosion govern drug release a simple kinetic model is unlikely to explain the overall in vivo release behavior. A level A correlation was established for the formulation with predominant diffusion controlled release. A level B correlation, however, was achieved even when drug release occurred by a combination of diffusion and erosion processes (64).
The ability of an IVIVC established with racemate metoprolol as well as each individual enantiomer in predicting the in vivo enantiomers performance has been recently investigated. Metoprolol racemate tablets with varying release rate (fast, medium, and slow) were used to perform the study. The result of the study indicated that IVIVC developed using R and S enantiomers were a good predictive of the R and S pharmacokinetic parameters, respectively. Racemate IVIVC was able to predict S enantiomer pharmacokinetic profile with the maximum prediction error of less than 5%, but not the R-enantiomer. This indicates that racemate data could not accurately predict R enantiomer concentrations. However, the racemate IVIVC was predictive of the active S stereoisomer (83). In another study, the correlation between the in vivo and in vitro drug release of R- and S-enantiomers of zileuton, a clinically effective agent in asthma, has also been attempted (54).
The lack of correlation between in vivo parameters and in vitro drug release data might be due to different reasons pertaining to the fundamentals of the process of study design, the complexity of the drug absorption, the weakness of the dissolution design, or for other reasons (84, 85).
NON-LINEAR CORRELATION
Both IVIVR (in vitro-in vivo relationship) and IVIVC are used to describe the relationship between in vitro dissolution data and in vivo pharmacokinetics. IVIVR is a more general term which allows the broad range of activities involved in relating in vitro dissolution to in vivo absorption and non-linear relationships. The most simple and appropriate relationships to consider first is the linear relationships, nonetheless non-linear correlations, even if uncommon, might be appropriate. IVIVCs reported in the literature are predominantly based on a linear relationship between the bioavailability parameters and in vitro release data (4, 36,37,54,56, 59, 60, 79, 83, 86). Non-linear (37, 87, 62) though predictive IVIVCs studies, however, are very scarce in the literature (87). In the IVIVC study reported by Sirisuth et al (87), linear and non-linear (quadratic, cubic and sigmoid functions) correlation models were examined using pooled fraction dissolved and absorbed from various combinations of the diltiazem extended release formulations. Plasma drug concentration profiles were predicted by convolution of the in vivo dissolution rates and the validity of the correlation was estimated by calculating prediction errors for Cmax and AUC for each formulation. Although the developed non-linear relationship permitted the dissolution data to adequately predict the bioavailability profile, the average prediction error observed for Cmax was 12.4% (2.4% greater than acceptable limits) and consequently the author concluded that the predictability of the quadratic IVIVC model, which provided a significant relationship, is inconclusive, and accordingly, should be externally validated. The failure of IVIVC predictability was attributed to high inter-subject variability which in turn might be due to inter-individual differences in first pass elimination (87).
Non-linear relationships between fraction dissolved and fraction absorbed was also observed with four series of extended release products containing ketoprofen. Modified isotonic phosphate buffer was used as dissolution medium and all four extended release products were incorporated in correlation (62). The data were fitted to the non-linear model proposed by Polli et al (88) using following equation:
![]() (15)
(15)
where, α is the ratio of a first order permeation rate constant to the first order dissolution rate constant, Finf, is the fraction of the dose absorbed at time infinity, and D, is a fraction of the total amount of drug absorbed at time t. For high values of α, dissolution is rate limiting step in absorption process and a linear level A IVIVC will be obtained. Small values of α give rise to a sort of parabolic relationship, similar to what is observed in this study, and indicates that in vitro drug release is initially more rapid than that of in vivo. With a coefficient of determination equal to 0.982, the value of α obtained by non-linear least square fitting was 1.92 consistent with an in vivo rapid initial dissolution rate as compared to that of in vivo. This parabolic non-linear relationship between fraction absorbed and dissolved may be the result of delayed in vivo dissolution which arises from gastric emptying and lower solubility of drug in an acidic environment as the drug is an acid with a pKa of 4.6 (62).
Linear and non-linear regressions have also been attempted for in vivo input and in vitro release for the controlled-release ethylcellulose-coated pellets containing adenosine derivative (34). The relationship between fraction absorbed in vivo and fraction released in vitro for membrane coated pellets was curvilinear indicating that there was a time-scale difference between in vivo and in vitro testing being much shorter for in vivo absorption. The authors, therefore, concluded that an in vitro dissolution test with a shorter time frame and faster release may be required to establish a linear IVIV correlation (34).
CONCLUSION
IVIVC includes in vivo relevance to in vitro dissolution specifications and can serve as surrogate for in vivo bioavailability and to support biowaivers. Furthermore, IVIVC can also allow setting and validating of more meaningful dissolution methods and specifications. It can also assist in quality control for certain scale-up and post-approval changes. Both the regulatory agencies and pharmaceutical industries have, therefore, understood this value of IVIVCs. Therefore, the activity in the area of IVIVC for oral extended release dosage forms has increased. The FDA Guidance on IVIVC provides general methods and guidelines for the establishment of IVIVC. The number of studies reported in the area of establishing IVIVCs for non-oral dosage forms are very scarce and further research is necessary in the development of more meaningful dissolution and permeation methods.