J Pharm Pharmaceut Sci (www.cspscanada.org) 9(3):388-397, 2006
Endoglin expression in hyper-cholesterolemia and after atorvastatin treatment in apoE-deficient mice
Nada Pospisilova1, Vladimir Semecky1, Gabriela Jamborova1, Katerina Pospechova1 , Dagmar Solichova2, Ctirad Andrys3, Petr Zdansky2 and Petr Nachtigal1
1Department of Biological and Medical Sciences, Faculty of Pharmacy, Charles University, Hradec Kralove, Czech Republic; 2Department of Metabolic Care and Gerontology, Medical School and Teaching Hospital, Charles University, Hradec Kralove, Czech Republic; 3Institute of Clinical Immunology and Allergology, Medical School and Teaching Hospital, Charles University, Hradec Kralove, Czech Republic.
Received Septemeber 4, 2006; Revised November 27, 2006; Accepted December 11; 2006, Published December 15, 2006
Correspondending Author: Dr. Petr Nachtigal, Department of Biological and Medical Sciences, Faculty of Pharmacy, Charles University Hradec Kralove, Heyrovskeho Czech Republic; Email: nachti@faf.cuni.cz
ABSTRACT -- Purpose. Endoglin (CD105) is a marker of activated endothelium and a modulator of TGF-β signaling. We hypothesized whether endothelial expression of endoglin is changed in hypercholesterolemia as well as whether its expression is affected by atorvastatin treatment in apoE-deficient mice. Methods. ApoE-deficient mice were fed with the chow diet for either 4 weeks or for 12 weeks respectively. In two treated groups, mice were fed with chow diet except atorvastatin was added to the diet for the last 4 weeks or for the last 8 weeks respectively, before euthanasia. Results. Administration of atorvastatin did not affect lipid parameters after 4 weeks treatment, however increased all lipid parameters after 8 weeks of treatment. Stereological analysis of immunohistochemical staining revealed that atorvastatin significantly decreased endoglin expression in endothelium after 4 weeks of treatment but increased it after 8 weeks of treatment. Conclusions. This study demonstrate that endoglin is expressed by aortic endothelium showing similar staining patterns like other markers involved in the process of atherosclerosis. In addition, we showed that endoglin expression in endothelium could be affected by the administration of atorvastatin beyond its lipid lowering effects in apoE-deficient mice.
INTRODUCTION
Atherosclerosis is a complex process that is characterized by the accumulation of modified low-density lipoprotein (LDL), local inflammatory and immune responses, and reduced nitric oxide bioavailability within the arterial wall (1). A central concept with regard to pathogenesis of atherosclerosis is that of endothelial cell dysfunction, which is associated with the release of a large number of mediators secreted predominantly by endothelial cells and leukocytes (2).
Transforming growth factor – beta (TGF-β), a widely expressed cytokine, is produced by both inflammatory and vascular cells and expressed in human and mouse atherosclerotic plaques (3). TGF-β exert their function through binding to a large family of specific receptors, including receptors type I, II, betaglycan, and endoglin (4). Among these, the serine-threonine kinase receptors types I and II are necessary for all tested biological responses to TGF-β and transmit the signal to downstream substrates through their kinase activity. By contrast, endoglin has been postulated as a regulator of TGF-β access to the signaling receptors (5).
Endoglin (CD105) is a homodimeric transmembrane glycoprotein composed of disulfide-linked subunits of 95 kDa. The primary sequence of human endoglin is composed of an extracellular domain of 561 amino acids, a single transmembrane region, and a cytoplasmic tail (6).
The major sources of CD 105 are vascular endothelial cells. Other cell types including vascular smooth muscle cells (7), fibroblasts (8), macrophages (9), leukemic cells of pre-B and myelomonocytic origin (10), and erythroid precursors (11) express CD105 to a lesser extent. Moreover endoglin is highly expressed in endothelial cells in tissues undergoing angiogenesis such as healing wounds, infarcts and in a wide range of tumors (12). In addition its expression was upregulated in human atherosclerotic plaques in the majority of smooth muscle cells (13). Lipid-lowering drugs offer one of the most effective therapeutic approaches used in clinical practice for the prevention and treatment of atherosclerosis (14). Statins, a well known class of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors, are active in the primary and secondary prevention of coronary heart disease and are the drugs most widely used for these purposes (15). A growing data base suggests that the beneficial actions of statins may be due to direct effects on the vascular wall in addition to lipid lowering. For instance, statins improve endothelial dysfunction via increased expression of nitric oxide (NO) and decreased expression of cell adhesion molecules in various animal models (16; 17).
Several papers provide evidence that TGF-β plays a major protective role in atherosclerosis (18). Moreover, as mentioned above, it has been demonstrated that endoglin is a modulator of TGF-β signaling and therefore it might affect these antiatherosclerotic effects of TGF-β. In addition, endoglin is a marker of activated endothelium (19; 20).
To the best to our knowledge nothing is known about the expression of endoglin in hypercholesterolemic conditions. Thus, in this study we wanted to evaluate the changes of endoglin expression in endothelium in very early stages of atherogenesis which comprise endothelial dysfunction. Moreover we hypothesized whether its expression is affected by an HMG Co-A reductase inhibitor atorvastatin. To test this hypothesis we examined the expression pattern of endoglin in endothelium in apoE-deficient mice by means of immunohistochemistry and stereology.
MATERIALS AND METHODS
The Ethical Committee of the Faculty of Pharmacy, Charles University, approved the protocols of the animal experiments. The protocol of experiments was pursued in accordance with the directive of the Ministry of Education of the Czech Republic (No. 311/1997).
Experimental Animals
Male homozygous apoE-deficient (apoE-/-) mice on a C57BL/6J background (n=32) weighing 15-20 grams were kindly provided by Prof. Poledne (IKEM, Prague, Czech Republic) and housed in the SEMED, (Prague, Czech Republic).
Experimental design
All mice were weaned at 4 weeks of age and randomly subdivided into four groups. Male apoE-deficient mice (n=8/group) were fed with the standard laboratory diet (chow diet) for either 4 weeks (non-treated apoE-/- 8 weeks) or for 12 weeks (non-treated apoE-/- 16 weeks ) respectively, after the weaning. In the two atorvastatin treated groups, mice were fed with the standard laboratory diet, except atorvastatin was added to the diet at the dosage of 10 mg/kg per day for the last 4 weeks (ATV apoE-/- 8 weeks ) or for the last 8 weeks (ATV apoE-/- 16 weeks) respectively, before euthanasia.
Each mouse in the atorvastatin groups lived in a separate cage obtaining 6g of food (in especially prepared pellets) daily with water ad libitum throughout the study. The food consumption was monitored every day. No differences in the food consumption were visible neither among animals of one experimental group nor between experimental groups. The dose of atorvastatin used in the present study was based on the doses used in previous studies with hyperlipidemic mice (21; 22).
At the end of the treatment period, all animals were fasted overnight and euthanized. Blood samples were collected via cardiac puncture at the time of death. The aortas, attached to the top half of the heart, were removed and then immersed in OCT (Optimal Cutting Temperature) embedding medium (Leica, Prague, Czech Republic), snap frozen in liquid nitrogen cooled 2-methylbutane and stored at ‑80°C until further analysis.
Biochemistry
Serum lipoprotein fractions were prepared using NaCl density gradient ultracentrifugation (Beckman TL 100, Palo Alto, CA). The lipoprotein fractions were distinguished in the following density ranges: very low density lipoprotein (VLDL) < 1.006 g/ml; low density lipoprotein (LDL) < 1.063 g/ml; high density lipoprotein (HDL) > 1.063 g/ml. Total concentration and lipoprotein fraction concentration of cholesterol were assessed enzymatically by conventional diagnostic kits (Lachema, Brno, Czech Republic) and spectrophotometric analysis (cholesterol at 510 nm, triglycerides, at 540 nm wavelength), (ULTROSPECT III, Pharmacia LKB Biotechnology, Uppsala, Sweden).
Immunohistochemistry
Sequential tissue sectioning started in the mouse heart until the aortic root containing semilunar valves together with the aorta appeared. From this point on, serial cross-sections (7μm) were cut on a cryostat and placed on gelatin-coated slides. Sections were air-dried and then slides were fixed for 20 minutes in acetone at -20°C. Endogenous peroxidase activity was blocked with 3% hydrogen peroxide in phosphate buffered saline (PBS) for 15 minutes. After blocking of nonspecific binding sites with 10% normal goat serum (Sigma-Aldrich Chemie, Steinheim, Germany) in PBS solution (pH 7.4) for 30 min, slides were incubated with primary antibodies for 1 hour at room temperature. After a PBS rinse, the slides were developed with biotinylated goat-anti rat IgG antibody diluted 1:400 in the presence of 200 mg/mL normal mouse IgG. Antibody reactivity was detected using HRP-conjugated biotin-avidin complexes (Vector Laboratories, USA) and developed with diaminobenzidine tetrahydrochloride as substrate. Specificity of the immunostaining was assessed by staining with nonimmune isotype-matched immunoglobulins.
Primary antibodies included the following: monoclonal antibody Rat Anti-Mouse CD31 (PECAM-1) diluted 1:100, monoclonal antibody Rat Anti-Mouse CD105 (endoglin) diluted 1:50. All antibodies were purchased from BD Pharmingen (California, USA)
Quantitative analysis of the immunohistochemistry
Stereological methods for the estimation of immunohistochemical staining of endoglin, and PECAM-1 were used as previously described (23; 24). In brief, the systematic uniform random sampling and the principle of the point-counting method were used for the estimation (25). A total number of 50 consecutive serial cross sections were cut into 7μm thick slices, which gave us 0.350 mm lengths of the vessel called the reference volume. This reference volume comprises several sections of the vessel containing semilunar valves in aortic root, and several sections of aortic arch (ascending part of the aorta). A systematic uniform random sampling was used in the reference volume. The first section for each immunohistochemical staining was randomly positioned in the reference volume and then each tenth section was used, thus five sections for each staining were used for the stereological estimation. The point-counting method was used and more than 200 test points per vessel, hitting immunostaining, were counted for an appropriate estimation (26). The estimated area is then:
![]()
where the parameter “a” characterizes the test grid and “P” is the number of test points hitting either the atherosclerotic lesion or positive immunostaining.
The area of PECAM-1 expression was considered as a total area of intact endothelium. Thus, the area of endoglin expression indicate the percentage of activated endothelial cells calculated as
![]() ,
,
where “area (x)” is the area of endoglin, in the endothelium and “area (PECAM)” is the area of PECAM-1 expression in the endothelium.
Photo documentation and image digitizing from the microscope were performed with the Nikon Eclipse E2000 microscope, with a digital firewire camera Pixelink PL-A642 (Vitana Corp. Ottawa, Canada) and with image analysis software LUCIA version 5.0 (Laboratory Imaging, Prague, Czech Republic). Stereological analysis was performed with a PointGrid module of the ELLIPSE software (ViDiTo, Kosice, Slovakia).
STATISTICAL ANALYSIS
All values in the graphs are presented as a mean ± SEM of n=8 animals. Statistical significance in the differences between groups was assessed by unpaired t-test with the use of the SigmaStat software (version 3.0). P values of 0.05 or less were considered statistically significant.
RESULTS
Biochemical analysis
In the first place, we examined the changes in serum lipoprotein fractions in non-treated mice. Biochemical analysis surprisingly revealed that total cholesterol (21.62 ± 2.94 vs. 11.21 ± 0.75 mmol/l, P = 0,022), VLDL (17.28 ± 2.54 vs. 8.49 ± 0.65 mmol/l, P=0.003), LDL (3.92 ± 0.38 vs. 2.46 ± 0.30, P = 0.023), and HDL (0.42 ± 0.04 vs. 0.23 ± 0.05, P = 0.043) levels were significantly decreased in 16 weeks old mice when compared with 8 weeks old mice (Fig.1).
Four weeks atorvastatin treatment significantly decreased TAG levels in 8 weeks old mice when compared to non-treated mice (1.57 ± 0.09 vs. 0.97 ± 0.12 mmol/l, P =0.014) (Fig.1). On the contrary, other lipid parameters were not affected by atorvastatin in these mice.
Eight weeks atorvastatin treatment significantly increased levels of the total serum cholesterol (11.21 ± 0.75 vs. 17.51 ± 1.16 mmol/l, P = 0.005), VLDL (8.49 ± 0.65 vs. 14.33 ± 0.89 mmol/l, P = <0.001), LDL (2.46 ± 0.30 vs. 3.51 ± 0.30 mmol/l, P = 0.036) and HDL (0.23 ± 0.05 vs. 0.40 ± 0.04 mmol/l, P = 0.029) in 16 weeks old mice in comparison to the non-treated mice (Fig.1).
Immunohistochemical staining of endoglin in apoE- deficient mice
ApoE-deficient mice develop spontaneous hypercholesterolemia on a chow diet which can be potentiated by atherogenic diet (27). However, the experimental design of this study was made to observe the changes of endoglin expression in endothelium where no atherosclerotic lesions were found. Thus no atherogenic diet was used.
PECAM-1 expression was observed only in endothelial cells in all groups of mice and this antibody was used as standard for the detection of intact endothelium (data not shown).
The staining patterns of endoglin were similar in both experimental groups. The expression of endoglin was detected in the endothelium of aorta in aortic sinus and aortic arch. Moreover, strong staining was
visible in small vessel and capillaries in myocardium (data not shown).
Endoglin staining in aortic endothelium was lower in 8 weeks old mice treated with atorvastatin (Fig 2B) when compared with non-treated mice (Fig 2A). On the contrary atorvastatin treatment resulted in stronger expression of endoglin (Fig 2D) in 16 weeks old mice when compared with non-treated mice (Fig 2C).
Stereological analysis of endoglin expression in apoE-deficient mice
Quantitave stereological analysis of endoglin staining showed a significant decrease in its expression in non-treated 16 weeks old mice when compared with non-treated 8 weeks old mice (22.0 ± 4.5 vs. 6.6 ± 1.5 %, P=0.007) (Fig. 3).
Moreover we demonstrated a significant decrease of endoglin expression after 4 weeks administration of atorvastatin in comparison with non-treated 8 weeks old mice (22.0 ± 4.5 vs. 5.3 ± 1.2 %, P = 0.013) (Fig. 3).
By contrast 8 weeks atorvastatin treatment resulted in significant increase of the endoglin expression when compared to non-treated 16 weeks old mice (6.6 ± 1.5 vs. 23.5 ± 9.5 %, P = 0.021) (Fig. 3).
DISCUSSION
The novel findings of the present study is that endoglin is expressed by endothelium in aortic sinus and aortic arch in apoE-deficient mice, and moreover that its expression is affected by the atorvastatin treatment.
Endoglin, a homodimeric transmembrane glycoprotein, is a component of the TGF-β receptor complexes (28). The expression of endoglin is predominant in endothelial cells, macrophages, fibroblast, and medial SMCs (12). Moreover it has been demonstrated that endoglin expression is increased during angiogenesis, and tumor development (29). Furthermore, endoglin expression was upregulated in medial smooth muscle cells, and endothelial cells in advanced atherosclerotic lesions in porcine carotid artery (20).
Since nothing is known about endoglin expression in hypecholesterolemic conditions, we wanted to evaluate its staining patterns in very early stages of atherogenesis, namely endothelial dysfunction in apoE-deficient mouse model of atherosclerosis.
ApoE-deficient mouse is a well-established genetic mouse model of atherogenic hypercholesterolemia, which is similar to hyperlipoproteinemia type III in humans.
We found that total cholesterol, VLDL, LDL and HDL was significantly decreased in 16 weeks old mice when compared with 8 weeks old mice. We do not have explanation for this phenomenon. However this surprising decrease in cholesterol levels with age in apoE-deficient mice were demonstrated by other authors (30; 31).
Statins competitively inhibit HMG-CoA reductase, the enzyme that catalyzes the rate-limiting step in cholesterol biosynthesis.
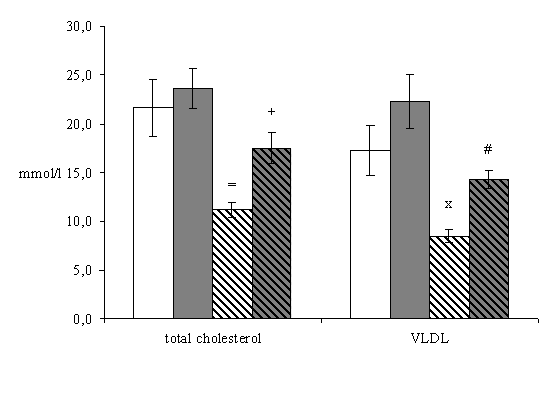
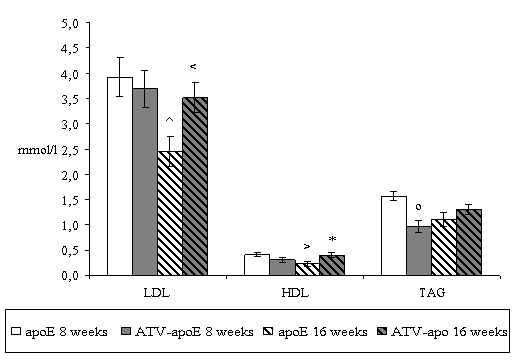
Figure 1. Serum lipid levels in apoE-deficient mice. Biochemical analysis revealed that in 16 weeks old non-treated mice there was significant decrease in levels of total cholesterol (=P = 0.022), VLDL (xP=0.003), LDL (^P = 0.023) and HDL (>P = 0.043) when compared with 8 weeks old non-treated mice. Four weeks atorvastatin treatment significantly decreases TAG levels in 8 weeks old mice (oP = 0.014) when compared with non-treated mice. On the contrary in 16 weeks old mice 8 weeks atorvastatin treatment significantly increases levels of the total cholesterol (+P = 0.005), VLDL (#P = <0.001), LDL (<P = 0.036) and HDL (*P = 0.029) when compared with non-treated mice. Male apoE-deficient mice (n=8/group) were used for the biochemical analysis.
......... 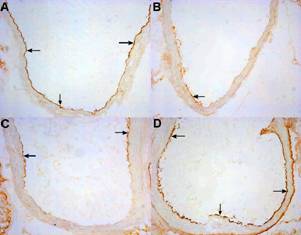
Figure 2. The figure demonstrates the intensity of immunohistochemical staining of endothelial expression of endoglin in non-treated and atorvastatin treated apoE-deficient mice. Endoglin staining (arrows) in aortic endothelium is lower in 8 weeks old mice treated with atorvastatin (B) when compared with non-treated mice (A). On the contrary atorvastatin treatment results in stronger expression of endoglin (arrows) (D) in 16 weeks old mice when compared with non-treated mice (C). Original magnification 200x.
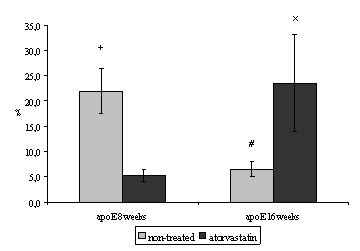
Figure 3. The percentage of activated endothelial cells for endoglin in both aortic root and aortic arch. In non-treated 16 weeks old mice the expression of endoglin is significantly decreased when compared with 8 weeks old mice (#P=0.0073). Endothelial expression of endoglin decreases after 4 weeks administration of atorvastatin in apoE-deficient mice (+P= 0.013 versus non-treated group). By contrast 8 weeks atorvastatin treatment results in a significant increase of the endoglin expression (xP = 0.021 versus non-treated mice. Male apoE-deficient mice (n=8/group) were used for the stereological analysis.
Moreover, recent experimental and clinical evidence indicate that some of the cholesterol-independent, or “pleiotropic” effects of statins involve improving or restoring endothelial function by enhancing the stability of atherosclerotic plaques, decreasing oxidative stress and inflammation, and inhibiting the thrombogenic response in the vascular wall (32).
We hypothesized, whether endothelial expression of endoglin is affected by atorvastatin treatment.
Endoglin expression was detected in myocardial capillaries and in endothelium of aortic sinus and aortic arch in all groups of mice showing similar staining pattern like cell adhesion molecules involved in the atherogenesis e.g. vascular cell adhesion molecule (VCAM-1) (33). Moreover, we demonstrated that 4 weeks administration of atorvastatin did not affect cholesterol levels in apoE-deficient mice, which is consistent with the results of Sparrow (22). However stereological analysis of immunohistochemical staining revealed that endothelial expression of endoglin was significantly lower in mice treated with atorvastatin. On the contrary 8 weeks atorvastatin treatment resulted in a paradoxical rise of all lipid parameters in apoE-deficient mouse. This effect of statin administration in apoE-deficient mice was previously described by other authors (30; 34; 35). This hypercholesterolemic effect of statins in apoE-deficient mice was recently elucidated by Fu et al., who demonstrated that enhanced hypercholesterolemia observed in apoE-deficient mice treated with statin is caused by alterations in the assembly of VLDL by the liver that contain more cholesterol than untreated mice (36). Moreover in our study, this hypercholesterolemic effect of atorvastatin was accompanied by the significant upregulation of endoglin expression in aortic endothelium in 16 weeks old mice. Thus, we suggest that hypercholesterolemic effect of atorvastatin overlay its direct effects on endothelium.
Taken together with the fact that decreased cholesterol levels in 16 weeks old non-treated mice was accompanied by the decrease in endoglin expression in endothelium when we compare it with 8 weeks old non-treated mice we might assume that the expression of endoglin could be affected by the changes of serum cholesterol in apoE-deficient mice. However, when we compare cholesterol levels and endoglin expression in 8 weeks and 16 weeks old atorvastatin treated mice we must state that this does not seem to be the case. Thus, cholesterol cannot be the only factor affecting the endothelial expression of endoglin. Therefore, we cannot exclude other factors e.g. proinflammatory state of endothelium that might be responsible for the increased endoglin expression in 16 weeks old atorvastatin treated mice.
As mentioned above, we showed that endoglin expression was reduced beyond lipid lowering effects of atorvastatin in 8 weeks old apoE-deficient mice.
It has been demonstrated that inhibition of HMG-CoA reductase by statins leads to the inhibition of nuclear transcription factor NF-kappaB which results in many beneficial pleiotropic effects of statins (37). Moreover, it has been demonstrated that multiple genes whose products are involved in the atherosclerotic process are regulated by NF-kappaB. This includes for instance E-selectin, monocyte chemoattractant protein-1 (MCP-1), and VCAM-1 (38). Furthermore, Rius et al. demonstrated that NF-kappaB consensus sequences found in the endoglin promoter might regulate endoglin transcription (39). Thus, it seems that the non- lipid lowering effect of atorvastatin on endoglin expression might be via an NF-kappaB dependent pathway. The same non-lipid lowering effect of atorvastatin on endothelial expression of VCAM-1 in apoE deficient mice was observed in our previous study suggesting both molecules might be regulated via an NF-kappaB dependent pathway (40)
TGF-β is a growth factor that exerts many regulatory actions. It is known for its role in development, proliferation, migration, differentiation, and extracellular matrix biology, but it is also an important immunomodulator (41). Moreover it has been demonstrated that endothelial cells and smooth muscle cells tend to be strongly inhibited by TGF-β both with respect to their proliferation and migration (42). In addition, macrophages and leukocytes are potently suppressed by TGF-β under most conditions tested (43). Thus, TGF-β is a strong anti-inflammatory agent that plays a protective role in the development of atherosclerosis (18). Since endoglin is a part of TGF-β receptor complex we might speculate that the expression of endoglin in the aorta could modulate the above mentioned TGF-β effects.
For instance it has been demonstrated that endoglin antagonizes the inhibitory effects of TGF-β and thus contributes to the proliferation, migration, and capillary formation of endothelial cells, the three key events in the angiogenic process (44). Moreover, endoglin is predominantly expressed in angiogenic endothelial cells and its expression is increased in tumor development. In addition endoglin expression was found to inhibit the TGF-β - dependent responses of cellular proliferation and PAI-1 expression (5).
If this inhibitory effect of endoglin would operate even for other effects of TGF-β, for instance inhibition of cell adhesion molecule expression (45) or vasodilatation, it is possible that increased expression of endoglin in hypercholesterolemia could results in inhibition of TGF-β signaling. In addition, this inhibition might result in the proinflammatory state in endothelium, increased activity of macrophages, T lymphocytes, thus actions that are necessary for the formation of atherosclerotic lesions.
In this study, we demonstrated that 4 weeks administration of atorvastatin decreases endothelial expression of endoglin. Thus, we assume that this decreased expression of endoglin by statin treatment might attenuate its inhibitory effects on TGF-β signaling which in turn could positively affect endothelial dysfunction in these mice.
On the other hand, increasing evidence indicates that endoglin may have functions independent of TGF-β. First, only a small percentage of surface-expressed endoglin actually binds TGF-β receptors (46). Second, despite the lack of signaling domains, endoglin overexpression affects cell morphology and adhesion in the absence of TGF-β (13). Thus, it is likely that endoglin cooperates with other different ligands. Thus, endoglin is a marker of activated endothelium regardless whether it affects TGF-β effects or not.
However, we must emphasize that this study was not designed to study possible interactions between TGF-β and endoglin. Therefore, further relation between endoglin and TGF-β in atherogenesis, as well as other effects of endoglin on markers of endothelial dysfunction for instance cell adhesion molecules expressions, such as NO production must be elucidated to reveal whether changes of endoglin expression could affect early atherogenesis.
There are some limitations of the study. First we used immunohistochemistry and stereology for the quantification of endoglin expression in endothelium in vessels where no atherosclerotic lesions were found. Thus, other studies focused on immunohistochemical and western blot analysis of endoglin expression in advanced atherosclerotic lesions must be made. Moreover, cell culture experiments with endothelial cells treated with or without atorvastatin in the presence or absence of cholesterol could confirm whether atorvastatin affects cholesterol-induced endoglin expression even in vitro.
In conclusion, this study demonstrate for the first time that endoglin is expressed by aortic endothelium showing similar staining patterns like other markers involved in the process of atherosclerosis. In addition, we showed that endoglin expression in endothelium could be affected by the administration of atorvastatin beyond its lipid lowering effects in apoE-deficient mice. Thus, prospective studies must be made to elucidate the role of endoglin and potential atorvastatin effects on its expression in more advanced atherosclerotic lesions and in relation to other markers of endothelial dysfunction and TGF-β signaling.
ACKNOWLEDGMENTS
The authors wish to thank Mrs. P. Jaburkova and Ing. Z. Mullerova for her skilful technical assistance, Mr. Robert Jaynes for the revision of the English text, Dr. Jiri Havranek, Zentiva, Czech Republic, kindly provided atorvastatin. This work was supported by the Grant Agency of the Czech Republic grant No. 304/03/P049, and by the Charles University Grant Agency Grant No. 101/2005C.