J Pharm Pharmaceut Sci (www.cspscanada.org) 9(3):317-326, 2006
Expanding the Scope of Crystal Form Evaluation in Pharmaceutical Science
Matthew L. Peterson, Magali B. Hickey, Michael J. Zaworotko and Örn Almarsson
TransForm Pharmaceuticals Inc. Lexington, MA, USA; Department of Chemistry University of South Florida, Tmpa, FL, USA
Received, May 31, 2006, revised, September 6, 2006; accepted October 4, 2006, Published November 1, 2006.
Corresponding Author: Dr. Matthew Peterson TransForm Pharmaceuticals, 29 Hartwell Avenue, Lexington, MA 02421 mpeterson@transformpharma.com
ABSTRACT -- The commentary seeks to provide a brief history and perspective on the importance of crystal forms of pharmaceuticals as a means of achieving performance criteria. The expanding scope of crystal form selection, emergence of crystal engineering in pharmaceutical science and pharmaceutical co-crystals are topics of this brief review.
THE ROLE OF MOLECULAR CRYSTALS IN PHARMACEUTICAL SCIENCE
Chemists and engineers in the pharmaceutical industry generally seek to deliver crystalline forms of their active compounds, mainly due to the inherent stability of crystalline materials and the well-established impact of crystallization processes on purification and isolation of chemical substances (1). Increasing attention is now being paid to the impact of materials properties in drug discovery and early development (2) as the drug compounds tend to be very valuable materials. The pharmaceutical industry’s mission for the material is to rapidly advance development programs with good confidence that form and formulation problems are unlikely to arise and to maximize a compounds’ potential as a therapeutic. This commentary seeks to raise the profile of crystal form studies and the emerging topic of crystal engineering in pharmaceutical science by discussing the “state-of-the-art” relating to pharmaceutical crystals and by expanding on possibilities that exist for future developments. In keeping with the common goal of making better products, faster and cheaper, we propose a paradigm of pro-active material design in pharmaceutical research. Rather than settling for the physical forms that the pure compounds intrinsically display, we should be aiming to identify the physical / material properties required of the target drug and make crystalline forms to meet those needs. We are not yet actively engaging in crystal engineering in the industry, and therefore we pose the question: How can we proceed toward making the crystal form that we want to use?
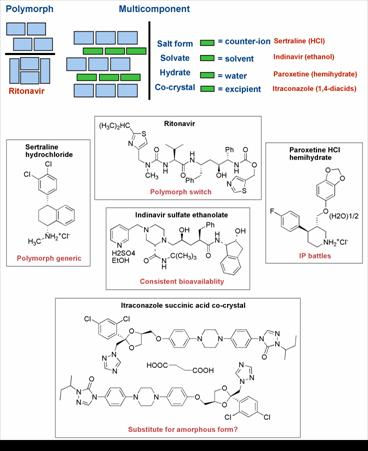
Figure 1. Examples of the types of crystal forms of pharmaceutical compounds used in products along with some issues and opportunities associated with each example
Polymorphism and pharmaceuticals
The phenomenon of crystal polymorphism, where the same chemical compound exists in more then one unique crystalline form, has been appreciated for over a century (3,4). Polymorphs of pharmaceuticals and drug candidates can occur in all types of phases (pure compounds, salts, solvates; for examples see Figure 1), though there is no way to predict the practical extent of polymorphism of any given compound (5). Based on recent reviews and commentaries, many strides have been taken towards better understanding of crystal polymorphism of pharmaceuticals (6,7,8).According to lore and some published examples, the puzzling and unpredictable phenomenon of crystal polymorphism has affected many projects in pharmaceutical research and development over the last few decades. Although most accounts of polymorphic transformation during product development remain anecdotal, there exist published examples of the problem from both early stage compounds (9) and marketed drugs (10).
The impact of crystal polymorphism and solvation state on pharmaceutical product value has been illustrated by costly product failures and protracted patent litigation examples. The former was most notably exemplified by the Norvir® capsule product failure in 1998, which was recounted and rationalized by solving the crystal structures of the Ritonavir polymorphs (10). In theory, all pharmaceuticals are vulnerable to such an unexpected and unlucky event which in the case of Ritonavir resulted in enormous cost and inconvenience for the innovator and impacted the patients’ use of this important HIV drug for roughly one year when no capsule formulation was available. Recently a new, meta-stable form of 5-fluorouracil was reported highlighting the potential for even very old drugs to unexpectedly reveal new polymorphs (11). On the litigation side, early entry of generic versions of some notable drugs has been enabled based on the use of a patently distinct but pharmaceutically equivalent polymorphs (or hydrates) (12). Ranitidine HCl (Zantac®) (5) and Paroxetine HCl hemihydrate (Paxil®) (13) are but two prominent examples where patent strategy and gaming created significant uncertainty about the value of a drug franchise. In response to these challenges, the pharmaceutical industry has developed processes and techniques for the identification and characterization of polymorphs and solvates of compounds of interest (6,14). Academics are contributing with both experimental and theoretical insights into crystal form study, prediction and engineering (7). Regulatory agencies also joined the effort in the 1990’s, as exemplified by the decision-tree approach to evaluating the impact of polymorphism on product performance (15,16). It seems fair to conclude that developments in the late 1980’s and many more during the 1990’s have brought the industry’s level of awareness, detection capability and decision making regarding polymorphs and solvates to new heights.
Beyond crystals and polymorphs
Another phase type of interest in pharmaceutical systems is the amorphous state of a compound. Amorphous drug preparations of small molecule drugs exist in the marketplace (17), and they usually represent deliberate efforts to avoid crystalline forms in order to meet a delivery objective. This is done when a crystalline form is unsuitable for oral absorption due to insufficient aqueous solubility and inadequate dissolution rate, particularly when particle size reduction does not ameliorate the impact of the latter to a satisfactory degree. Infrequently, however, no suitable crystalline forms of a compound can be found making the amorphous material the only viable option. Examples include itraconazole, quinapril HCl, zafirlukast, cilastatin and nelfinavir mesylate. In such cases, it would seem important to gain insights into the structural factors that lead to the outcome, in an effort to assess risk of eventual crystallization events. The characterization and application of amorphous pharmaceutical compounds and dispersions have been topics of review elsewhere, and will not be further discussed, given that the focus of the present contribution is on crystalline compounds (18-20).
Particle size reduction of poorly absorbed compounds is a proven approach to enhance biopharmaceutical performance. Milling to progressively smaller sizes can be considered a progressive approach towards amorphous, high-energy states. A special case of such crystal size modification is the advent of the nano-dispersion approach in pharmaceutical products. A key technology development in this regard is NanoCrystals® (Elan corp.), which has been successfully applied in three oral products to date: Rapamune®, Tricor® and Emend®. The latter two compounds are excellent examples of the impact of media milling and physically stabilizing a crystalline dispersion of nanometer dimensions (100’s of nm) particles with the ultimate goal of significant bioavailability improvement. The physical challenge is one of re-growth of particles (Ostwald ripening), which is addressed by the use of specific surface stabilizers in formulation development. The active ingredient in Tricor, fenofibrate, is a low-melting, oil soluble (but water-insoluble) substance. The bioavailability of this compound is increased by decreasing particle size, with the nano-dispersion being close to optimal. In addition, the NanoCrystal formulation of Tricor shows a decreased dependence of oral bioavailability on food compared to the previous formulation of Tricor (21) In the case of Emend, the solubility of the drug substance, aprepitant, is on the order of 0.3 mg/mL and the doses are in 100’s of mg/day. Oral bioavailability of this drug is a strong function of the particle size, with the smallest achievable crystals being optimal (22) The example illustrates that size modification is a viable strategy to be considered alongside form modification.
Salt selection – A powerful strategy for crystal form optimization
Pharmaceutical developers have focused efforts on finding and formulating a thermodynamically stable crystalline form with acceptable physical properties for a given compound. This is reasonable, given the need to avoid cascading from a meta-stable form to a more stable one in unpredictable fashion. Occasionally certain physical properties, such as low aqueous solubility, are limiting to performance of the compound, leading to poor oral bioavailability or insufficient solubility for an injection formulation. One of the main strategies used to affect physical performance of a compound and one that is often employed by pharmaceutical scientists is the practice of salt selection (23). At least half of compounds in marketed products are in the form of a salt for one reason or another. This fact alone speaks to the versatility of the salt selection approach. Salt forms of a pharmaceutical can have many benefits, such as improved stability characteristics, optimal bioavailability and aqueous solubility for an injectable formulation. Salts, like all other crystalline forms, are subject to polymorphism and solvate formation, thus requiring the same form identification studies as are needed for a neutral compound. A remarkable example of co-optimization of properties is indinavir (HIV protease inhibitor), which is marketed as the sulfate salt ethanol solvate (24,25) The crystalline free base has variable oral bioavailability in dogs (26,27) and humans (28). While acidic solutions of the base compound showed good oral pharmacokinetics, the stability of the drug in acidic solution is not consistent with a product (26). Therefore, the discovery of the salt form ensured both shelf stability and robust bioavailability performance.
The salt selection strategy is limited in two ways. First, salt formation relies on the presence of one or more ionizable functional groups in the molecule; many drugs and development compounds lack this feature. Second, our ability to predict a priori whether a given compound will form a crystalline salt (or salts) is non-existent. The ability to actively identify crystalline salt forms has been confined to manual empirical evaluation using multiple salt formers for a given acid or base. Recently advances have been made in the area of high-throughput salt selection and crystal engineering strategies associated with salt formation (14,29-32). In one case, we have advocated the simultaneous assessment of polymorphism as a way to help rank the developability of different crystalline salts (14). While salt forms will continue to have a prominent place in pharmaceutical science, the need for enhanced productivity dictates that every advantage must be sought to aid the design of an appropriate crystalline form of an active molecule. Specifically, the ability to design scaffolds into crystalline forms will enhance our capacity to convert interesting molecules into effective drugs. Crystal engineering offers some additional tools in this regard.
Crystal engineering and co-crystals
Crystal engineering is generally considered to be the design and growth of crystalline molecular solids with the aim of impacting material properties. A principal tool is the hydrogen bond, which is responsible for the majority of directed intermolecular interactions in molecular solids. Co-crystals are multi-component crystals based on hydrogen bonding interactions without the transfer of hydrogen ions to form salts – this is an important feature, since Brønsted acid-base chemistry is not a requirement for the formation of a co-crystal. Co-crystallization is a manifestation of directed self-assembly of different components. Co-crystals have been described of various organic substances over the years (33,34) and given various names, such as addition compounds (35,36) molecular complexes (37,38) and heteromolecular co-crystals (39). Regardless of naming convention, the essential meaning is that of a multi-component crystal where no covalent chemical modification of the constituents occurs as a result of the crystal formation. Pharmaceuticals co-crystals have only recently been discussed as useful materials for drug products.
Pharmaceutical co-crystals
Pharmaceutical co-crystals can be defined as crystalline materials comprised of an active pharmaceutical ingredient (API) and one or more unique co-crystal formers, which are solids at room temperature. Co-crystals can be constructed through several types of interaction, including hydrogen bonding, p-stacking, and van der Waals forces. Solvates and hydrates of the API are not considered to be co-crystals by this definition. However, co-crystals may include one or more solvent/water molecules in the crystal lattice.
An example of putative design, a construction and preparation process is shown in Figure 2 for the 5-fluororuracil:urea 1:1 co-crystal(40). This real example neatly illustrates the opportunity and challenge that exists currently with designing pharmaceutical co-crystals. Firstly, the ‘design’ is challenging because we have no ability to predict the exact crystal structure that may result from a crystallization attempt. By analogy to the challenge of deriving protein structure from first principles, the primary sequence (chemical structure in our case) is known and elements of secondary structure (the 2-D tape construction in Figure 2) are somewhat discernible from primary information. Prediction of the actual 3-D folded conformation (tertiary structure or obtained by self-assembly) is not possible. In other words, while we currently have the ability to project which things associate in what approximate manner on the secondary level, crystal structure prediction is essentially an intractable proposition. By extension, and just as the exact function of a protein and quantitative parameters of activity are not predictable from primary and secondary structure, the prediction of crystal properties is not possible in the absence of structural information and measurements.
There is early evidence that practitioners were aware that apparent co-crystallization of drugs could lead to useful preparations (41). In fact, a ‘chemical compound’ composed of sulfathiazole and proflavin dubbed flavazole was used to treat bacterial infection during the Second World War (42). The case of flavazole reveals insight into how two different molecules might interact in a putative co-crystal:“… flavazole is definitely a chemical compound containing equimolar proportions of sulphathiazole and proflavin base. It is believed that combination occurs through the acidic sulphonamide group (SO2NH) of the sulphathiazole and the basic centres of the proflavin. Perhaps the most realistic expression of the formula would be to place proflavin and sulphathiazole side by side with a comma between them.” (42) In the second half of the 20th century, interest in co-crystals evolved into the directed study of intermolecular interactions in crystalline solids (43-45). The technical development of routine single-crystal structure determination led to a watershed of data, now largely accessible through the Cambridge Structural Database (CSD) (46,47). The structural data have become useful for understanding the intermolecular interactions in co-crystals in atomic level detail (48). Using insight gained from analysis of the CSD and directed experimentation, scientists attempt design of co-crystals with specific properties, such as color or non-linear optical response, by selecting starting components with appropriate molecular properties likely to exhibit specific intermolecular interactions in a crystal (49-52). However, even when chemically compatible functional groups are present it is not possible to accurately predict if a co-crystal, a eutectic mixture or simply a physical mixture will result from any given experiment. As a result of these complexities, attention has been directed at the identification and characterization of intermolecular packing motifs with the goal of developing principles for co-crystal materials (53).

Figure 2. Steps involved in crystal engineering of a pharmaceutical phase, exemplified by the real example of co-crystallization of 5-fluorouracil and urea.
PROSPECTS FOR CRYSTAL ENGINEERING AND PHARMACEUTICAL CO-CRYSTALS
At the beginning of the 21st century, the field of crystal engineering has experienced significant development. Importantly, crystal engineering principles are now being actively considered for application to pharmaceuticals to modulate the properties of these valuable materials (54). Because the physical properties that influence the performance of pharmaceutical solids are reasonably well appreciated, there is a unique opportunity to apply crystal engineering techniques and the appropriate follow-up studies to solve real world problems, such as poor physical and chemical stability or inadequate dissolution for appropriate biopharmaceutical performance of an oral drug. As structures and series of pharmaceutical co-crystals have begun to appear, we again find that properties cannot be predicted from the structures. Nevertheless, occasional trends have been suggested. For example, insoluble drug compounds co-crystallized with highly water soluble complements tend to achieve kinetic solubilities in aqueous media several times greater than the pure form (55,56). There are also more possible phases for each given active compound to consider, thus there will arguably be a greater opportunities for property enhancement. In terms of stability enhancement and solubilization, the example of the series of itraconazole co-crystals with pharmaceutically acceptable 1,4-diacids (55) suggests a strategy alternative to amorphous drug formulation. The co-crystal options presented retain the stability inherent in a crystalline state, while allowing for solubilization that significantly exceeds that of crystalline itraconazole base and rivals the performance of the engineered amorphous bead formulation (Sporanox®).
Where are we now? From recent literature it appears that knowledge gained over the past century and increasingly sophisticated screening techniques developed within the last decade are paving the way towards design of co-crystals with potentially improved pharmaceutical properties (55-58) In terms of the application to pharmaceutical systems, the field of crystal engineering is developing the retro-synthetic understanding of crystal structure using reasoning that is analogous to that applied by organic chemists. For example, the retro-synthetic approach in covalent synthesis operates on the level of a single molecule, while the analogous effort in crystal engineering focuses on the “supermolecule”: The assemblies that define the crystalline arrangement of the molecules as they self-organize into the solid-state. The parallels between the development of crystal engineering and synthetic organic chemistry run still deeper. Methodologies for carrying out these crystallizations are being developed alongside the development of new robust motifs (6,53,55,57,60). The importance of the solubility and dissolution relationships of the components of a putative co-crystal is becoming a matter of significant investigation (56,60). The same can be said for the roles of additives in templating novel forms. Mechanical milling of materials has also been documented as a means to make co-crystals, and a recent example of polymorphic forms of caffeine:glutaric acid illustrates the opportunities of this type of processing to influence crystal form (61). With an increase in the understanding of the modes of self-assembly, one can start to address the design aspect towards making pharmaceutical co-crystals.
There remain several limitations to the application of what is currently known to the design of useful materials. As mentioned earlier, it remains intractable to reliably predict crystal structure. Multi-component crystals are well out of reach for prediction due in part to complex energetic landscapes, lack of appropriate charge density models and a large number of degrees of freedom, making computation unfeasible. Moreover, there is only a qualitative understanding of the interplay between intermolecular interactions and materials performance, especially for properties relevant to pharmaceuticals such as solubility, dissolution profile, hygroscopicity and melting point. But the saving grace of the co-crystal approach comes in two guises: Complementarity and diversity. On the topic of complementarity, it is possible, by way of CSD database mining for instance, to identify trends of hetero-synthon occurrence in model systems. As for the diversity aspect, the space of possible co-crystal formers is large, limited only by pharmaceutical acceptability. Coupled with parameters such as stoichiometry variation and increase in the number of components (binary systems can be expanded into ternary ones, etc.), the opportunities appear vast.
THE FUTURE OF CRYSTAL ENGINEERING IN PHARMACEUTICAL SCIENCE
Where are we going? At this point, we have only just scratched the surface of materials science-driven pharmaceutical product design. In the 21st century, practitioners of pharmaceutical chemistry need to enumerate and exploit the opportunities of crystal form design that nature affords us, and thus gain increasing ability to design the materials we need from the molecules that we seek to convert into pharmaceuticals. Learning will be facilitated by advances in crystallization automation (6,62), microscopy-spectroscopy techniques (Raman and IR microscopy) and new techniques such as terahertz spectroscopy and AFM, along with increasingly sophisticated X-ray diffraction lab instrumentation. In addition, further enhancements in the data mining tools associated with the CSD operating on an ever increasing number of high-quality crystal structures will undoubtedly lead to new knowledge and principles of interaction.
The challenge placed before pharmaceutical scientists, now and in the future, is the following: (i) to understand the requirement of a particular compound in terms of materials structure and properties, and (ii) to creatively integrate crystal engineering within the limits of pharmaceutical acceptability of components to obtain new forms of active ingredients with desirable properties for formulation and delivery. It should become the collective mantra of medicinal chemists, process engineers and pharmaceutical scientists to “design and make the material we need.” This mantra can form the common aspiration for an industry that is in significant need of innovation and productivity enhancement.
CONCLUSIOns
The rapidly evolving field of pharmaceutical crystal engineering is primed for a prominent position in the development and improvement of pharmaceutical products. Scientists in the pharmaceutical industry need to be attuned to the possibilities that the approach can offer. In the next few decades, this awareness needs to become general across the industry and among the disciplines that contribute to drug development. The expanding role of crystal engineering in pharmaceutical science is waiting to be filled with insights and experience from past work in other areas, stimulated by present challenges in the interest of developing the drugs of the future.