J Pharm Pharmaceut Sci (www.cspscanada.org) 9(3):271-280, 2006
Evaluation of synthetic polymeric micelles as a stabilization medium for the handling of membrane proteins in pharmaceutical drug discovery
Olga V. Trubetskoy, Moshe Finel, Thomas J. Burke and Vladimir S. Trubetskoy
Quintessence Biosciences, Inc., Madison, WI, USA; Department of Pharmacy, University of Helsinki, Finland
Received March 1, 2006; Revised received June 24, 2006; Accepted July 31, 2006, Published, August 25, 2006
Corresponding Author: Dr. V. S. Trubetskoy, Quintessence Biosciences, Inc., 505 S. Rosa Rd., Madison, WI 53719, USA, Email: vladimirt@quintbio.com
ABSTRACT: PURPOSE Polymeric micelles have been used for solubilization of insoluble drugs and as carriers for drug delivery applications. Here we evaluated an application of the synthetic polymeric micelles in experiments designed to improve the handling and stability of membrane proteins targets. METHODS: Particle sizing by dynamic light scattering was performed in a Zeta Plus Photon Correlation Spectrometer at 532 nm. UGT1A1 activity has been measured in fluorescent assay using scopoletin as a substrate. COX-2 activity has been measured in a fluorescent assay using Amplex Red. Fluorescence Resonance Energy Transfer (FRET) was monitored using either 463 nm excitation wavelength (the emission range 500–600 nm) or 395 nm excitation wavelength (the emission range 500–600 nm). RESULTS: Incorporation of membrane proteins into PreserveX™-QML polymeric micelles resulted in improved homogeneity and stability of the preparation and in reduced light scattering. Stabilization of the biological activity of micelle-incorporated membrane proteins, such as the human UGT1A1 and COX-2 both during extended incubations at room temperature and during multiple freeze/thaw cycles, has been achieved. CONCLUSION: PreserveX™-QML polymeric micelles help to homogenize and disperse membrane proteins preparations and stabilize the biological activity of the proteins making it more suitable for pharmaceutical assays and applications.
Introduction
Polymeric micelles are particulate self-assemblies in aqueous media that are composed of linear amphiphilic macromolecules possessing both hydrophilic and hydrophobic ‘blocks’ (AB-type) on a single strand (each copolymer strand is amphiphilic). At the appropriate ratio of block lengths, these copolymers spontaneously form spherical particles in water: the hydrophobic blocks form the ‘core’, while the hydrophilic blocks form the surrounding “corona” (Figure 1). The particle sizes range between 10–100 nm, making them considerably smaller than phospholipid vesicles (liposomes) (1). Another distinction of polymeric micelles is the absence of internal aqueous space. Like other micelle-forming amphiphiles, AB-type copolymers will dissociate upon dilution below their CMC. However, due to the polymeric nature of the hydrophobic blocks, polymeric micelles possess very low CMC values and are thermodynamically and kinetically quite stable. Typical CMCs for polymeric micelles lie in the low micromolar range, whereas the CMCs for many commonly used low-molecular weight surfactants are in the millimolar or high micromolar range (2). Such low CMCs indicate that polymeric micelles require an energy input to dissociate and incorporate other materials, like membrane components.
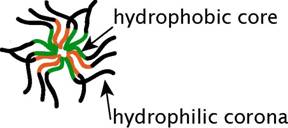
Figure 1. Schematic of a polymeric micelle
Polymeric micelles have many potential uses in biomedicine. For example, polymeric micelles have been proposed for use in intraparenteral drug delivery applications, especially for delivery of drugs with low aqueous solubility (3) or as carriers for protein/peptide therapeutics (4). In drug discovery research, polymeric micelles offer promise due to their ability to maintain the structure and activity of membrane proteins under the conditions required for HTS assays or structure/function studies. Researchers have developed numerous varied formulations of amphiphilic polymeric preparations for stabilizing membrane proteins.
Abbreviations:
|
These approaches include hydrophobized low molecular weight poly(acrylic acid) derivatives (amphipols) that successfully stabilized bacteriorhodopsin, the bacterial photosynthetic reaction center, and cytochrome b5, hydrophobically modified natural polymers (amphiphilic carboxymethylpullulans or amphibiopols) that effectively solubilized and stabilized membrane proteins containing either α-helix or β-sheet domains from the outer membranes of Pseudomonas fluorescens (6), so-called peptitergents composed of amphiphilic alpha helices (7), lipopeptides (8), and poly(ethyleneglycol)-lipid (PEG-lipid) conjugates (9). However, each approach has its limitations. For example, amphibiopols have a high anionic net charge (6), making them unsuitable for some membrane proteins. The PEG-lipid conjugates are not very efficient solubilizers, are negatively charged, and tend to form amphiphile-stabilized bilayer disks of widely varying size (9). Moreover, simply preparing the components of these systems can be a non-trivial task (10). Here we evaluated commercially available preparations of polymeric micelles, PreserveX™-QML, for their ability to support and stabilize activity of the micelle-incorporated membrane proteins targets.
Membrane proteins, including receptors, transporters, ion channels, and a variety of enzymes, facilitate many of the cell’s most basic functions of metabolism, growth, and death. Research into membrane proteins has contributed to our understanding of almost all cellular operations including transcription, translation, cellcycle control, division, secretion, signal transduction, and cellular architecture. Inherited and acquired abnormalities in these proteins are involved in numerous human diseases and disorders (11, 12) including cystic fibrosis (13), cancer (14-16), diabetes (17), and Alzheimer’s disease (18, 19). Despite their pharmaceutical importance, structures have been determined for only some 1% of the known membrane proteins, because of the unique challenges they present (20, 21). Recombinantly produced human membrane proteins are now the major source of membrane proteins used in pharmaceutical industry. In most cases, such proteins are produced as heterogeneous mixtures of membrane-containing subcellular fractions derived from the host cells. Due to their heterogeneity and large particle size, these fractions tend to precipitate out of solution during handling and storage, resulting in protein aggregation, denaturation, and the production of significant light scattering that can interfere with many downstream applications (22). The inability to produce stable, active preparations of membrane proteins exhibiting a reduced light scattering presents major problems for pharmaceutical drug discovery and is a roadblock for structural characterization and for developing assays for membrane protein targets (23-25).
The addition of low molecular weight detergents to preparations of membrane protein targets may help to reduce heterogeneity and light scattering, but often leads to instability and loss of activity. The stabilization mechanism of membrane proteins by the detergents (surfactants) is not fully understood. The overall influence of a surfactant on transmembrane protein domains may be the sum of two opposing factors: enthalpy, which favors association, and entropy, which favors dissociation (26). Hence, employing an appropriate amphiphile, with both hydrophobic and hydrophilic regions, might lead to a robust membrane protein-stabilizing effect. This hypothesis has been experimentally confirmed, with strong evidence indicating that some surfactants can indeed stabilize transmembrane protein domains (27-29). Typically, membrane proteins tend to aggregate and precipitate when the concentration of free surfactant falls below its CMC. While maintaining the concentration of free surfactant above its CMC can improve the stability of membrane proteins, high concentrations of low molecular weight surfactant often destabilize membrane proteins (24).
Due to stability and light scattering problems, developing assays that employ membrane protein targets represent a formidable challenge in pharmaceutical drug discovery. A combination of low biochemical activity, light-scattering effects, and low stability make membrane proteins one of the most intractable targets for assay development and structural analyses (20, 30). In general, membrane-bound proteins will remain stable as concentrated lipid suspensions for several hours at room temperature. However, they rapidly inactivate when diluted to working concentrations (1–10 µg/ml). For example, cytochrome P450 (CYP450) preparations are known for their limited stability (31); incubation of key CYP450 isoforms at 25°C for six hours reduced their activity by about 90% (32). Other drug metabolizing enzymes, such as membrane-bound UDP-glycosyltransferases (UGTs), are even more labile (33). Stability problems have also been reported for other membrane proteins, such as cyclooxygenases (34). Microsomal preparations of the recombinantly expressed membrane proteins serve as an important tool in pharmaceutical drug discovery. However, these preparations are often difficult to handle in various assays and applications due to their heterogeneity resulting in protein aggregation and precipitation out of solution and inactivation during multiple freeze-thaw cycles and interference with fluorescence-based assay technologies. Here we evaluated an amphiphilic polymeric preparation of PreserveX™-QML as a novel approach to improve handling and stability of the selected membrane proteins preparations used in pharmaceutical drug discovery.
Materials and METHODS
Reagents
PreserveX™-QML was obtained from QBI Life Sciences (Madison, WI). The membranes containing recombinant UDP-glucuronosyl transferase 1A1 (UGT1A1) and α2-adrenergic receptor fused with green fluorescent protein (α2-AR-GFP) were prepared as described in (35) and (36), respectively. The fluorescent phospholipid labels used in the FRET experiments were N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanol-amine (NBD-PE) Lissamine™ rhodamine B 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (Rh-PE), both obtained from Invitrogen (Eugene, OR). Black 96-well microtiter plates were purchased from Corning Life Sciences (Corning, NY). Membrane fractions from Sf9 cells containing recombinantly expressed human motilin receptor (hMTLR) and DIPYrromethene BOron Difluoride (BODIPY)-labeled motilin used in the fluorescence polarization experiments were purchased from PerkinElmer Life and Analytical Sciences, Inc. (Boston, MA). Unlabeled human/porcine motilin was obtained from Phoenix Pharmaceuticals (Belmont, CA). All other reagents and chemicals were purchased from Sigma/Aldrich (St. Louis, MO).
Fluorescence resonance energy transfer (FRET) experiments with fluorescently labeled lipids and proteins
PreserveX™-QML was labeled with fluorescent lipids using the following procedure. PreserveX™-QML reagent (9.9 µl) was lyophilized overnight in a 1.5-ml microcentrifuge tube and dissolved in 100 µl chloroform. NBD-PE (11 µg) and Rh-PE (5 µg) were both added to the dissolved polymer solution. The polymer/fluorescent lipid mixture was dried, first under a stream of N2 gas, and second in high vacuum for 2 h. The mixture was reconstituted in 0.5 ml HEPES-buffered saline (HBS) (20 mM HEPES, 150 mM NaCl, pH 7.5). A Safire fluorescence plate reader (Tecan US, Durham, NC) and black 96-well microtiter plates (Corning) were used for both sets of experiments. In both experiments, control preparations were prepared as described above, but the sonication step was omitted. For the FRET experiments with lipid labels, the UGT1A1 membranes (50 µg total protein) were mixed with PreserveX™-QML labeled with NBD-PE and Rh-PE (3 µg total polymer) in 100 µl HBS. The mixture was sonicated for 30 s in a bath-type sonicator and dispensed into black 96-well microtiter plates (100 µl/well). FRET was monitored using a 463 nm excitation wavelength (the emission range was 500–600 nm). For the lipid/protein FRET experiments, 40 µg of α2-AR-GFP-containing membranes were mixed with 10 µg of Rh-PE-labeled PreserveX™-QML (2 mol % Rh-PE) in 100 µl HBS and sonicated for 30 s in a bath-type sonicator. The excitation wavelength was 395 nm (for GFP) (the emission range was 500–600 nm).
Extraction procedure
To allow PreserveX™-QML to interact with natural membranes to form mixed micelles, PreserveX™-QML and fluorescently labeled membranes were combined in a 1.5-ml microtube in 0.5 ml HBS (total volume) at the appropriate protein/polymer ratio and briefly vortexed. If necessary, the mixture was sonicated briefly with a bath-type laboratory sonicator (VWR Model 75) at the maximum power setting (9) for 30 s at room temperature to complete dispersion. Immediately after sonication, the mixed micelle solution was dispensed into black 96-well microtiter plates (100 µl/well) for (Corning) fluorescence measurements.
Dynamic Light Scattering
Particle sizing by dynamic light scattering was performed in a Zeta Plus Photon Correlation Spectrometer equipped with a 50-MW solid-state laser emitting at 532 nm (Brookhaven Instruments Corp., Holtsville, NY). The concentrations of UGT1A1 membranes and QML micelles were 0.2 mg/ml and 4 mg/ml, respectively, in 0.5 ml of filter-sterilized HBS (0.22-µm filter). The correlation curves were collected in logonormal mode. Particle size distribution data were calculated using Brookhaven Instruments Zeta Plus Particle Sizing software.
UGT assay
We measured the UGT1A1 activity following modifications to the procedure of Broudy and co-workers (37) using scopoletin as a substrate for glucuronidation. Briefly, scopoletin and uridine 5΄-diphosphoglucuronic acid (UDPGA) were mixed in HBS at 20 µM and 1 mM, respectively and then dispensed in black 96-well microtiter plates (Corning) (100 µl/well). UGT1A1-containing membranes (2.5–10 µl) were added to each well to initiate the assay. Enzyme activity was monitored by following the decrease in scopoletin fluorescence at λex = 410 nm and λem = 590 nm using a Safire fluorescence plate reader (Tecan US). Control preparations were prepared as above, but addition of UDPGA was omitted.
Motilin receptor assay
Fluorescence polarization (FP) measures molecular rotation occurring during the fluorescence lifetime, the period between the excitation of the fluorophore and its subsequent light emission. Because small molecules tumble quickly in solution, when they are excited by plane-polarized light, the light these molecules emit is relatively depolarized. Larger molecules and complexes, however, tumble more slowly in solution. Therefore, when they are excited by plane-polarized light, the light these molecules or complexes emit remains more highly polarized (38). The FP displacement assay we used involves the addition of unlabeled motilin to a reaction mixture containing BODIPY-labeled motilin bound to either PreserveX™-QML-incorporated hMTLR or native hMTLR. As the concentration of unlabeled motilin increases, the unlabeled motilin displaces the BODIPY-labeled motilin. The free BODIPY-labeled motilin is a much smaller molecule than the motilin-hMTLR complex and, therefore, displacing BODIPY-labeled motilin decreases the polarization value. The experiments were performed in 20 mM HEPES buffer, pH 7.5, containing 10 mM MgCl, 22.5 µg/ml hMTLR membranes, 5 nM BODIPY-labeled human motilin, and 1 mg/ml PreserveX™-QML in a total volume of 100 µl. The fluorescence signal was measured using a Tecan Ultra fluorescence polarization plate reader (Tecan US) with a rhodamine fluorescence polarization filter. We conducted the control reactions identically, except that no PreserveX™-QML was added to the control samples.
RESULTS
Ability of PreserveX™-QML to incorporate fluorescent labels and to form mixed micelles with lipid and protein components of biological membranes. Monitoring the disappearance of FRET in liposomes following dilution with unlabeled vesicles has been used previously to determine the ‘dilution’ of the original membrane after treatment with excess of unlabeled vesicles and to demonstrate liposomal membrane fusion (39). We employed FRET to demonstrate lipid/protein intermixing with PreserveX™-QML. For this experiment, we used a commonly used established FRET pair comprising of two fluorescent lipids, 7-nitrobenz-2-oxa-1,3-diazol-4-yl phosphatidylethanolamine (NBD-PE) and rhodamine-phosphatidylethanolamine (Rh-PE), that were incorporated into PreserveX™-QML at 1% (mol). Upon excitation at 463 nm (the excitation wavelength for NBD-PE), we observed an intense emission signal at 580 nm (the emission wavelength for rhodamine), indicating that FRET has occurred (Figure 2). After addition of 17-fold excess of UGT1A1 membranes (w/w) and brief sonication, the signal at 580 nm disappeared, indicating that the two fluorescent lipids were no longer in close proximity and that FRET had been disrupted.
Once the fluorescent labels were incorporated into PreserveX™-QML, they could be intermixed not only with the lipid components of biological membranes, but also with the protein components. To demonstrate this, we employed α2-AR-GFP-containing membranes (carrying the α2-adrenergic receptor fused with green fluorescent protein) (36) and PreserveX™-QML labeled with Rh-PE. GFP and rhodamine are a known FRET pair (40). After brief sonication of the PreserveX™-QML/membrane mixture, we detected a significant FRET signal at 585 nm, indicating efficient intermixing of PreserveX™-QML with the membrane components (Figure 3).
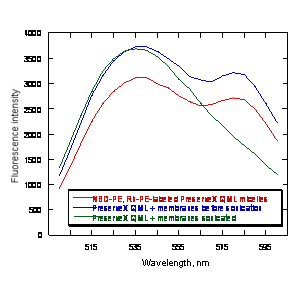
Figure 2. Transfer of PreserveX™-QML-incorporated lipophilic fluorescent labels to the PreserveX™-QML-incorporated biological membranes. The decrease in observed FRET between PreserveX™-QML-incorporated NBD-PE and Rh-PE after co-sonication with biological membranes indicates that the fluorescently labeled PreserveX™-QML has incorporated membrane components, thereby disrupting FRET.
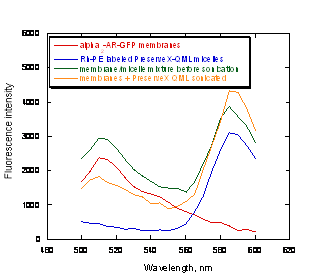
Figure 3. FRET signal increases after α2-AR-GFP fusion protein is co-sonicated with Rh-PE-labeled PreserveX™-QML.
Role of mild sonication in membrane dispersion with PreserveX™-QML
Polymeric micelles are thermodynamically and kinetically stable particles (3). In our PreserveX™-QML/biological membrane intermixing experiments, a detectable FRET signal appeared only after the mixtures were sonicated (41) in a bath-type sonicator. The resultant mixed polymeric micelles exhibited critical micelle concentrations (CMC) two to three orders of magnitudes lower than those observed with commonly used surfactants, such as CHAPS (3).
In addition, incorporating PreserveX™-QML into biological membranes led to a significant reduction in particle size. Figure 4 illustrates that the average particle size measured using dynamic light scattering of the sonicated PreserveX/membrane complexes was more than ten-fold smaller compared to the original microsomal membrane preparations. Significant narrowing of particle size distribution has also been observed (Figure 4). Hence, although PreserveX™-QML incorporates both the lipid and protein components of membranes into mixed polymeric micelles, the overall result very closely resembles solubilization of natural biological membranes.
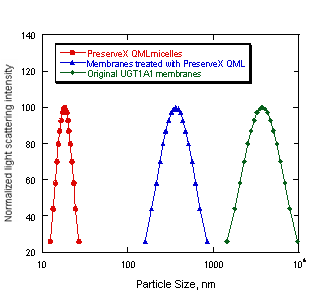
Figure 4. Changes in particle size after co-sonication of UGT1A1 biological membranes with PreserveX™-QML. The sonication was carried out as outlined in Materials and Methods section. The data represent averaged particle size distributions calculated in unimodal regimes using dynamic light scattering spectrometer.

Figure 5. PreserveX™-QML-stabilized UGT1A1 enzyme activity increases after multiple freeze/thaw cycles in HBS.
Enzyme activation after multiple freeze/thaw cycles
We have compared the relative activity of UGT1A1, incorporated into PreserveX™-QML micelles or stored in HBS buffer, during multiple freeze/thaw cycles. As Figure 5 demonstrates, the enzyme activity of PreserveX™-QML-stabilized UGT1A1 actually increased after three freeze/thaw cycles. In contrast, the activity of the UGT1A1 samples stored in buffer remained unaltered. Increased activity of UGT1A1 after multiple freeze-thaw cycles in a presence of polymeric micelles is a phenomenon closely related to a latency of UGT enzymes where an increase of enzyme activity has been observed after addition of the membrane-modifying agents to microsomal preparations (42, 43).
Competitive displacement FP assays performed with and without PreserveX™-QML
For pharmaceutical drug discovery studies, it is critical to establish that incorporating membrane proteins into a new amphiphilic medium does not alter their quantitative biological properties (such as binding characteristics). Therefore, we have performed fluorescence polarization-based (FP-based) competitive displacement assays with PreserveX™-QML-incorporated and native forms of human motilin receptor (hMTLR), using wild-type A9L cell membranes as a control. Motilin receptors belong to the family of Class I G-protein-coupled receptors that are involved in regulating gastro-intestinal motility (44). As Figure 6 demonstrates, the assays showed that both native and PreserveX-incorporated forms of hMTLR exhibited similar competitive displacement curves for BODIPY-motilin with non-labeled motilin, thus proving that the presence of PreserveX™-QML did not interfere with the ligand binding characteristics of this membrane protein.
DISCUSSION
We have examined the performance of PreserveX™-QML polymeric micelles in multiple experiments designed to test its ability to incorporate membrane components and fluorescent labels enabling their use to homogenize membrane preparations and to stabilize and support protein activity under different conditions and in fluorescence-based assays for pharmaceutical drug discovery. As Figure 2 shows, we have demonstrated that PreserveX™-QML can incorporate lipophilic fluorescent labels and that labeled PreserveX™-QML can be intermixed with the lipid and protein components of biological membranes.
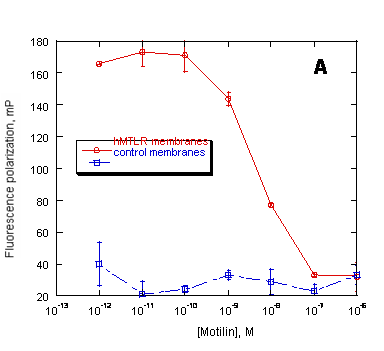
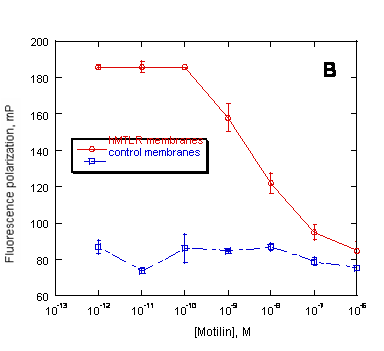
Figure 6. Preservation of ligand binding characteristics of PreserveX™-QML-incorporated hMTLR measured in a fluorescence polarization (FP) assay. FP measured the displacement of BODIPY-labeled motilin by unlabeled motilin binding to (A) native hMTLR membranes and (B) PreserveX™-QML-incorporated hMTLR (hMTLR membranes). Wild-type A9L cell membranes (PerkinElmer) were used as control membranes.
Here, incorporating membrane components disrupted the FRET between a dye pair that had been incorporated into PreserveX™-QML, as shown by the decreased signal intensity at the 585-nm peak. As Figure 3 demonstrates, the presence of membrane components in PreserveX™-QML did not interrupt FRET. In this experiment, we combined Rh-PE-labeled PreserveX™-QML with α2-AR-GFP-containing membranes. Separately, Rh-PE-labeled PreserveX™-QML and α2-AR-GFP-containing membranes exhibited characteristic fluorescence signals for Rh-PE and GFP, respectively. After mixing, the signals from both dyes remained intense; no FRET occurred. Following sonication, however, when the Rh-PE-labeled PreserveX™-QML and the α2-AR-GFP fusion protein were incorporated into mixed polymeric micelles, FRET occurred: the signal from the 515-nm GFP peak decreased while that from the 585-nm Rh-PE peak increased. This result indicates that the α2-AR-GFP fusion protein was in close proximity to Rh-PE and, therefore, demonstrates that the α2-AR-GFP fusion protein was incorporated into Rh-PE-labeled PreserveX™-QML.
Light scattering poses a significant problem for fluorescence-based assays. Because membrane protein preparations produce relatively large particles of varying sizes, they are generally not well suited for fluorescence-based assays. As Figure 4 illustrates, however, the size of mixed PreserveX™-QML-membrane component polymeric micelles were not only much smaller than the particles in most membrane preparations, but the size range was also much narrower. As a result, PreserveX™-QML is well suited for fluorescence-based assays.
Another long-standing problem with membrane proteins is their overall instability during storage. Many cannot withstand more than a few freeze/thaw cycles without losing their activity. In the example shown in Figure 5, the activity of UGT1A1 stored in buffer did not change, while the activity of PreserveX™-QML-incorporated UGT1A1 actually increased by about 30%, during three freeze/thaw cycles. One possible explanation for this phenomenon is activation of the UGT enzyme during multiple freeze-thaw cycles in a presence of PreserveX™-QML. Latency in activity has been reported for several members of UGT families with increased enzyme activity reported upon addition of pore-making peptides, such as alamethicin (42, 43). Pore-making peptides can provide access to additional substrate and cofactor binding sites on the UGT molecule, thus increasing enzyme activity. Exposing UGT1A1-containing membrane preparations to multiple freeze/thaw cycles in the presence of PreserveX™-QML may similarly affect UGT by unmasking additional substrate and cofactor binding sites, resulting in increased specific activity of the enzyme preparations. Similar activation in a presence of PreserveX™-QML has been also observed for other classes of membrane proteins, such as cyclooxygenases and membrane-associated receptor tyrosine kinases (unpublished observations). Although not all membrane proteins will experience such increased activity, these results demonstrate that PreserveX™-QML can be successfully used to boost and preserve activity of some membrane proteins during storage and multiple freeze-thaw cycles.
Many membrane proteins require the presence of associated lipids or cofactors to retain their native conformation and activity. Because detergent-solubilized membrane proteins are separated from these components, they often lose activity. In contrast, PreserveX™-QML incorporates both the lipid and protein components from membranes and does not separate protein subunits. As a result, membrane proteins incorporated into PreserveX™-QML retain their activity. Figure 6 proves that PreserveX™-QML-incorporated hMTLR fully retained the ability to bind its ligand, motilin. In other experiments (data not shown), we have demonstrated that PreserveX™-QML-incorporated membrane proteins retain their activity during extended room-temperature incubations far better than the native membrane protein preparations.
Conclusions
We have evaluated PreserveX™-QML polymeric micelles as a new approach for incorporation, stabilization and developing pharmaceutical assays for membrane proteins targets. These polymeric micelles can readily incorporate membrane components and lipophilic dyes. Membrane proteins preparations incorporated into such micelles are well-suited for fluorescence-based assays due to their small, uniform particle size and significantly reduced light scattering. Equally important, the incorporation into polymeric micelles can stabilize the biological activity of selected membrane proteins, both during extended incubations at room temperature under dilute assay conditions and during multiple freeze/thaw cycles. PreserveX™-QML can enable drug discovery researchers to more easily investigate important membrane proteins. However, application of the PreserveX™-QML does not automatically ensure solubilization and retention of functional properties for all classes of membrane proteins and may not be suitable for some. For others, the composition of the correct solubilization and stabilization media will have to be established experimentally by optimization of the medium conditions, such as pH, ionic strength, or the presence of lyotropic salts, and other protective ligands such as glycerol and sucrose (2).