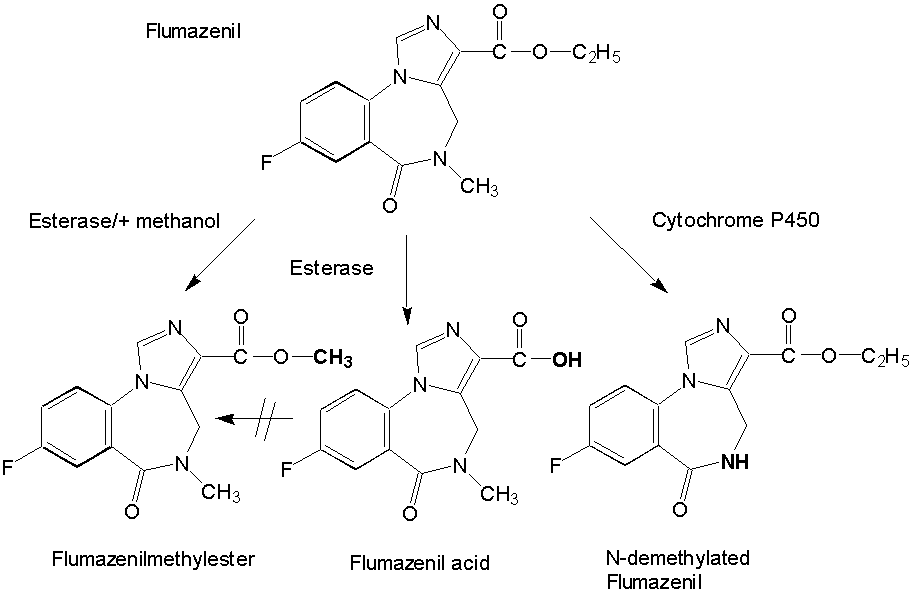
Isolation and Pharmacological Characterization of Microsomal Human Liver Flumazenil Carboxylesterase Britta Kleingeist, Ronald Böcker, Gerd Geisslinger and Roland Brugger1, Department of Experimental and Clinical Pharmacology and Toxicology, University of Erlangen-Nürnberg, Universitätsstr. 22, D-91054 Erlangen, Germany Abstract Purpose. In vivo the biotransformation of the imidazobenzodiazepine antagonist flumazenil leads to the formation of two metabolites, flumazenil acid and N-demethylated flumazenil. In the present study we investigated the role of carboxylesterases for the metabolism of flumazenil. Methods. We purified a non-specific carboxylesterase (EC 3.1.1.1) from human liver microsomes that catalyzes the hydrolysis of flumazenil to flumazenil acid and, in presence of methanol the formation of flumazenil methyl ester an in vivo unknown metabolite. The purification procedure included solubilization of the microsomes obtained from human livers with Triton X-100 and subsequent chromatography of the 100 000 x g supernatant on blue-sepharose, DEAE-sepharose, hydroxyapatite and final chromatofocusing. Results. The purified esterase isozyme exhibited an apparent subunit molecular weight of 59 kDa as estimated by SDS gelelectrophoresis, a native molecular weight of 170 kDa determined by a calibrated gel filtration column suggesting that the active enzyme is a trimer. The isoelectric point of the enzyme was approximately 5.4. The specific activities of the purified enzyme were 5.8 nmol/(min*mg protein) protein for the formation of flumazenil acid and 31 nmol/(min*mg protein) for the synthesis of the flumazenil methylester. The purified enzyme obeys simple Michaelis-Menten kinetics with KM values of 665 mM for flumazenil acid, 1011 mM for methanol and 900 mM for the flumazenil methylester. PMSF, a specific inhibitor for serine proteases and mammalian acetylcholinesterase, completely inhibited the formation of flumazenil -acid and the flumazenil methylester at a concentration of 100 mµM. No synthesis of the flumazenil -methylester could be observed by incubation of the purified esterase with flumazenil acid in the presence of methanol leading to the conclusion that the enzymatically catalyzed reaction is a transesterification. The purified esterase was digested with endoproteinase LysC. A 15 amino acid long peptide was isolated and showed identical matches to carboxylesterase cDNAs from human liver and lung. Conclusion. Our results show that carboxylesterase isozymes play an important role in the detoxification and metabolism of flumazenil. Because of enzymatic, catalytic and structural properties a similarity of the characterized flumazenil carboxylesterase with human liver cocaine carboxylesterase is possible. Abbrevations: DTT, dithiothreitol; SDS, sodiumdodecylsulfat; PAGE, polyacrylamid gelelectrophoresis; PMSF, phenylmethylsulfonyl fluoride; IEF, isoelectricfocusing; FA, flumazenil acid; MF, flumazenil methylester. Introduction The imidazobenzodiazepine-derivative flumazenil is one of several 1,4-benzodiazepine derivatives with high affinity for the benzodiazepine receptor that act as competitive antagonists (1, 2). It is the only benzodiazepine receptor antagonist available for clinical use at present and blocks many of the actions of benzodiazepines but does not antagonize the CNS effects of other sedative-hypnotics, ethanol, opioids, or general anesthetics (3). Flumazenil is used also as a diagnostic drug in benzodiazepine intoxication (4). Flumazenil binds specifically to benzodiazepine receptors in the brain blocking the behavioural, neurological and electrophysiological effects of several benzodiazepines, but lacks the pharmacological effects of classical benzodiazepines. Analyzing human plasma flumazenil was inactivated by hydrolysis to flumazenil -acid (FA) and probably by cytochrome P450 catalyzed N-dealkylation to N-demethylated flumazenil (N-DE), the major urinary metabolite of the drug (Figure 1).
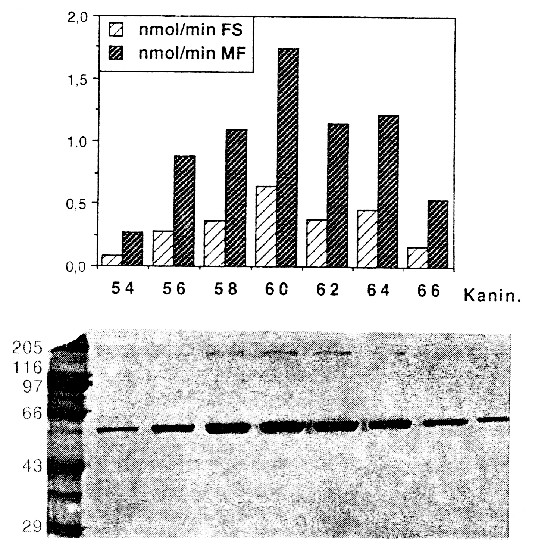
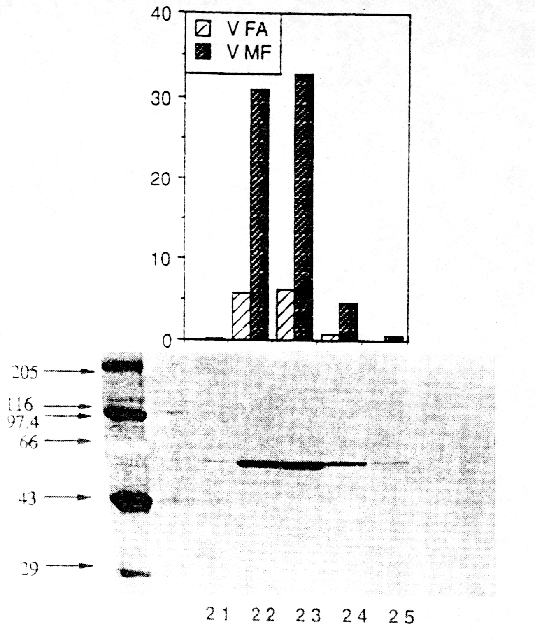
In urine and plasma from humans (5, 6), dogs and rats the two metabolites were formed in varying quantities (7). It is unknown which metabolites are formed exclusively in the liver and which enzymes are responsible for the ester cleavage and for the synthesis of the N-demethylation product. Previous studies have shown that the benzodiazepines midazolam and diazepam are metabolized by cytochrome P-450 enzymes (8, 9). Because of the similar chemical structures of midazolam and flumazenil, it was supposed, that the formation of the N-demethylated product and possibly the ester cleavage are catalyzed by cytochrome P-450 enzymes as known for the ester cleavage of other drugs (10). Recently we reported on the in vitro metabolism of flumazenil in human liver microsomes and suggested that microsomal liver carboxylesterases are the key enzymes in the metabolism of flumazenil to flumazenil acid (11, 12). Here, we report on the purification and characterization of a human liver flumazenil carboxylesterase that catalyzes both the formation of flumazenil acid from flumazenil and the formation of flumazenil methylester from methanol and flumazenil (Figure 1). This enzyme may play a key role in regulating tissue levels of flumazenil and its metabolites. Materials and MethodsChemicals All reagents and solvents were of highest quality available. Flumazenil, flumazenil acid and N-demethylated flumazenil were kindly provided by Hoffmann La-Roche, Basel, Switzerland. Purified rabbit- and pig carboxylesterase (EC 3.1.1.1.) were purchased from Sigma (Deisenhofen, Germany) and Boehringer (Mannheim, Germany). Acetonitrile was from Fisons (Loughborough, England). Glucose 6-phosphate, glucose 6-phosphate deydrogenase, and NADP+ were obtained from Boehringer (Mannheim, Germany). Other chemicals were obtained from Merck (Darmstadt, Germany). Chromatography materials Blue-Sepharose CL-6B fast flow, DEAE-Sepharose, Mono P HR 5/5, Polybuffer 94, PBE 74 and Sephadex G-25 were obtained from Pharmacia LKB (Freiburg, Germany) and hydroxyapatite from Biorad (München, Germany). Centriplus-30 concentrators were purchased from Amicon (Witten, Germany). Human liver samples Human liver samples were from patients (Department of Clinical Pathology, by courtesy of Prof. Dr. C. Wittekind) undergoing liver resection for various clinical reasons. After histomorphological examination the remaining tissue, which was not used for further tests, was immediately frozen in liquid nitrogen and stored at -70 °C until preparation of the microsomal fraction. Purfication of human liver flumazenil carboxylesterase Human liver microsomes were prepared from 60 g of frozen human liver by homogenisation in a buffer containing sucrose (0.25 M), Tris-base (5 mM), EDTA (1 mM, pH 7.4) and subsequent differential ultracentrifugation as described by Brugger et al. (13). The microsomes were resuspended in buffer (50 ml) containing K2HPO4 (20 mM), DTT (2 mM), EDTA (1 mM, pH 7.4). Solubilization of the microsomes was achieved by adding dropwise Triton X-100 (20 mL, 0.6% v/v, final concentration 0.3% v/v, protein concentration approximately 1 mg/mL) and stirring the protein suspension (1 h) on ice. Then the mixture was centrifuged (100 000 x g, 1h 15 min). The resulting supernatant (60 mL) was collected and diluted with 5 volumes of a solution containing K2HPO4 (20 mM), DTT (2 mM), Triton X-100 (0.15 % v/v, pH 7.4; buffer A). The diluted solution (300 mL) was applied to a Blue-Sepharose column (50 mL gel, fast flow quality, XK 26/20 Pharmacia, Freiburg, Germany) equilibrated with buffer A. The protein containing the activity for flumazenil acid formation did not bind on the blue-sepharose material. The eluate were directly applied on a DEAE-sepharose column equilibrated with buffer A and then eluted with a linear concentration gradient established between buffer A (60 mL) and the same volume of buffer A containing 200 mM NaCl. The protein fractions containing activity for flumazenil acid formation (15 mL) were combined, concentrated to 2 mL with Centriplus 30 (Amicon, Witten, Germany) and diluted with five volumes of buffer A and applied on a hydroxyapatite column (XK 16/20 Pharmacia, Freiburg, Germany). The column was washed with buffer A (3 volumes) and then eluted with a linear concentration gradient established between buffer A (60 mL) and the same volume of buffer A containing potassium phosphate (200 mM). The flow rate was 0.5 mL/min and fractions of 2.5 mL were collected. The enzyme with the flumazenil acid and flumazenil -methylester activity appeared in a single peak, which coincided with the main protein peak. The fractions exhibiting high activities and a protein band at 59 kDa in the SDS gel were pooled (30 mL) and concentrated to 2 ml with Centriplus 50 (Amicon, Witten, Germany). After concentrating the pool of active fractions, the buffer was exchanged for 25 mM bis- Tris pH 7.1. The sample was applied to a Polybuffer Exchanger chromatofocusing column (Mono P HR 5/5). The enzyme exhibiting both activities appeared in a single peak. Bio-Rad protein assay (Bio-Rad, München ,Germany) based on the Bradford dye-binding procedure (14) was used for measuring total protein concentration with bovine serum albumin as standard. SDS polyacrylamide gel electrophoresis with minigels (10 x 9 cm) containing 10% polyacrylamide was performed with a system from GIBCO BRL (Mini-V 8.10) according to the procedure of Laemmli (15). After electrophoresis the gels were silver stained (16). Determination of peptide sequence We determined the partial amino acid sequence of esterase using the protein purified by classical chromatography. Because the protein was found to contain a blocked N-terminal, 30 mµg of the homogenous protein were fractionated by SDS-PAGE and transferred to a PVDF-membrane. After visualization with Coomassie staining the 59 kD band was excised and digested with 1 mg endoproteinase Lys-C in 400 ml digestion-buffer (0.1 M Tris-HCl, pH 8.0, 0.2 mM CaCl2, 10% CH3CN, 1% NP40) at 24 °C for 8 hr. The resulted peptide mixture was fractionated by Superspher 60 RP select B (Merck, Darmstadt) reversed phase HPLC. One dominant peak fraction from the RP-HPLC chromatography were loaded on a sequencer (Beckman PI 3000) and analyzed (Toplap, Gesellschaft für Angewandte Biotechnologie mbH, München, Germany). The peptide sequence was compared with entries in the SWISS protein sequence data base using the Protein Sequence Analysis System (International Biotechnologies, Inc., New Haven, USA). Determination of flumazenil acid and flumazenil methylester synthesis 100 mg protein was incubated at 37 °C in 0.1 M KH2PO4 buffer (pH 7.4) with flumazenil (25-3000 µM) or acetylsalicylic -acid (25-3000 µM, a marker substrate for carboxylesterases) in a total volume of 0.5 mL. Flumazenil was dissolved in methanol (final concentration 1% of the solvent). The reaction was stopped by the addition of 50 µL of H3PO4 (43%, w/w) and 1.5 µg oxazepam were added as an internal standard. Incubations were extracted with 1.6 mL CH2Cl2 and the organic layers evaporated to dryness by a stream of nitrogen. HPLC-Analysis Residues were dissolved in 200 µL of a CH3CN/H2O mixture (1:1 v/v) and 50 µL aliquots were injected for HPLC analysis. The products of flumazenil metabolism were analyzed on a Spectra-Physics system (Thermo Separation Products, Darmstadt, Germany) with a SP 8800 ternary-pump and a Spectra Focus optical scanning detector. Aliquots of the samples were injected by a Gilson Model 232 Bio autosampler (Abimed, Langenfeld, Germany) and analyzed on an ET 250/8/4 Nucleosil® 7 C18 equipped with a Nucleosil® 120-5 C8 11x4 mm I.D. guard column (Macherey-Nagel, Düren, Germany). The eluent consisted of CH3CN/0.5 % H3PO4 (38 %/62 % v/v) at a flow rate of 1mL/min. The metabolites were monitored by UV-detection: 245 nm (flumazenil and metabolites), 235 nm (acetylsalicylic -acid and salicylic-acid). Inhibition studies Inhibition by chemicals was tested by coincubation of inhibitors and flumazenil (concentration 500 mµM) with microsomes for 30 min at 37°C. Analyses of the products were carried out as described above. Data analysis For the determination of KM and Vmax values of flumazenil acid and flumazenil methylester formation, 400 µg protein were incubated with different flumazenil concentrations (between 125-3000 µM) diluted in 50% methanol/water. For the determination of the kinetic parameters for methanol 200 µg protein were incubated with 500 µM flumazenil in the presence of different methanol concentrations (124-2472 mM) for 30 min at 37 °C. The KM and Vmax values for the formation of flumazenil -acid and and flumazenil -methylester from flumazenil and for methanol were obtained from Eadie-Hofstee plots of the data (17). Results A carboxylesterase that hydrolyzes flumazenil to flumazenil acid and catalyzes the methyl transesterification of flumazenil with methanol to form flumazenil methyl ester was isolated from human liver microsomes by a five-step purification procedure.Non-specific esterase activity (p-nitrophenol forming), flumazenil esterase activity and methyl ester transferase activity were measured by HPLC at each step of the isolation procedure. Both flumazenil esterase and methyl transferase activity eluted together with approximately the same ratio of activities in the various protein peaks and fractions measured (Figures 2 and 3). Gel electrophoresis of fractions with higher activities showed a strong correlation between the amount of enzyme at 59 kDa and the determined activities (Figures 2 and 3). An increase of specific activity was observed using either flumazenil or p-nitrophenyl acetate (not shown) as substrate for the enzyme assay.
The yield of flumazenil esterase was 9%, and a 116-fold purification was achieved for the flumazenil esterase activity. The enrichment of the methyl transferase activity was 155 fold and therefore in good correlation to the esterase activity (Table 1). Table 1. Purification of microsomal human liver flumazenil carboxylesterase: measurement of products formed by incubation of protein fraction with flumazenil in the presence of methanol. FA, flumazenil acid; MF, flumazenil methylester; Units are defined as nmol of product/min.
The purity of the enzyme preparation was examined by SDS-PAGE (Figure 3) and IEF (not shown). A single protein was observed on SDS gels (Figure 3). The subunit molecular weight of 59 kDa for the protein was estimated from the migration distance using a standard curve generated with high range SDS-PAGE markers. A gel filtration on a Superose 6 column revealed a molecular weight of approximately 170 kDa for the native protein, suggesting that the active enzyme is a trimer. These data are similar to previously reported results about non specific carboxylesterases from rat (18, 19) and pig (20). The protein stain on the IEF from the pure flumazenil carboxylesterase sample showed a series of closely spaced bands may be due to glycosylation, which is a known feature of the esterase protein family. An isoelectric point of approximately 5.4 was determined. To characterize the primary structure of the esterase, the purified protein was digested with LysC at pH 8.0. Peptides were separated by RP-HPLC and sequenced by automated Edman degradation. The first sequenced peptide with a length of 15 amino acids (NH 2-Glu-Gly-Tyr-Leu-Gln-Ile-Gly-Ala-Asn-Thr-Gln-Ala-Ala-Gln-Lys-COOH) perfectly matched (100% identity) the deduced sequence for carboxylesterase cDNAs from human liver (21) and human alveolar macrophaghes (22).Incubation of purified carboxylesterase with flumazenil in the presence of methanol resulted in the detection of two metabolites: flumazenil acid and flumazenil methylester. The resulting flumazenil methylester was identified and characterized by GC/MS analyses. HPLC purified flumazenil methyl ester exhibited characteristic mass fragments: m/z 289 indicating the molecular weight, m/z 257 resulting from the ester cleavage -O-CH 3 and m/z 229 -COO-CH3. For steady state kinetic analysis, the rate of flumazenil acid and flumazenil methylester formation was measured at different substrate concentrations of flumazenil and data were estimated from Eadie-Hofstee plots (not shown). The apparent KM for flumazenil degradation to flumazenil acid was 665 ± 122 µmM, tofor flumazenil-methylester 900 ± 68 µmM (mean ± SD) and 101 ± 10 mM for methanol. Initial rates for flumazenil acid and flumazenil methylester formation remained linear for 30 min of incubation and for protein concentrations up to 0.8 mg/mL. Incubation of commercially available rabbit esterase revealed a KM of 180 ±14 µmM and 2,1 ± 0,19 mM for flumazenil acid and the methylester formation, respectively. For pig carboxlesterase the KM values were 141 ± 15 µmM and 1,45 ± 0,13 mM for flumazenil acid and flumazenil methylester production, respectively (Table 2).Table 2. KM values and 1Vmax ( nmol/(mg protein x min)) for flumazenil degradation to flumazenil acid and to flumazenil methylester determined for carboxylesterases from different species. FA, flumazenil acid; MF, flumazenil methylester; n.d. not determined.
Incubation of flumazenil acid, dissolved in methanol, with human liver microsomes or the purified enzyme did not result in the production of flumazenil methylester suggesting that flumazenil methylester is only formed by transesterification. This transesterification of flumazenil occurred faster than the hydrolysis, because 2.8 times more methylester than flumazenil acid was formed. PMSF an inhibitor of serine proteases and mammalian acetylcholinesterase inhibited the metabolism of flumazenil to flumazenil acid and flumazenil methylester. In human liver homogenateT the formation of flumazenil acid was completely inhibited at a concentration of 100 µM PMSF and the remaining activity for flumazenil methylester formation was 8.6 ± 1.9 % indicating an additional enzyme catalyzing the flumazenil transesterification to flumazenil methylester. N-benzimidazole (NBI) is described as an excellent inhibitor of the most abundant cytochrome P-450 enzymes. Incubation of the purified carboxylesterase with NBI (1 mM final concentration) and flumazenil (dissolved in methanol) in the presence of a NADPH-generating system did not inhibit the metabolism of flumazenil to flumazenil acid and flumazenil methylester indicating that the enzyme which catalyzes the synthesis of flumazenil acid and methylester is not P450 like. Discussion In vivo the biotransformation of a drug can occur by spontaneous, noncatalyzed chemical reactions, but the majority of reactions are catalyzed by specific cellular enzymes. For example the wellknown cytochrome P450 superfamily is responsible for most of the oxidation and reduction reactions, while other enzymes like esterases and amidases catalyze the hydrolysis of ester-, thioester- and amide-bonds in the metabolism of a variety of endogenous substrates and exogenous xenobiotics. Carboxylesterases have a broad substrate specificity and beside their physiological functions they play an important role in the detoxification of xenobiotics (23). It is suggested that these enzymes metabolize lipophilic compounds to more hydrophilic carboxylic acids or alcohols (24). Multiple isozymes of the superfamily carboxylesterases have been isolated from livers of rat, pig, rabbit and human. The isozymes were differentiated according to the isoelectric point, substrate specifity, and inhibitor specifity. Most of these enzymes are characterized as microsomal glycoproteins (25, 26, 27). Recently we investigated the metabolism of flumazenil in human liver microsomes and suggested that a carboxylesterase from human liver microsomes is involved in the degradation and metabolism of flumazenil (11, 12). While different human carboxylesterases have been described, we here reported on the isolation and characterization of a human carboxylesterase that catalyzes the hydrolysis of flumazenil to flumazenil acid and in vitro the transesterification of flumazenil to flumazenil methylester. In line with our previous results, we have characterized the flumazenil carboxylesterase by determination of enzymatic, catalytical and structural properties. The identity of a peptide sequence obtained after isolation and fragmentation of the flumazenil carboxylesterase with the deduced sequence for carboxylesterase from human liver (20) and human alveolar macrophages (21) and taking into account the previously published high correlation of the formation of salicylic acid (from acetylsalicylic acid) and the flumazenil acid formation (from flumazenil), rule out that the ester cleavage of flumazenil is catalyzed by a cytochrome P-450 enzyme as known for other drugs (10). On the other hand the identity of human liver cocaine carboxylesterase (28) with the cDNA reported from Munger et al. (21) for human alveolar macrophages may implicate that this human carboxylesterase has a broader substrate specificity as suggested so far. In addition cocaine carboxylesterase catalyzed the formation of cocaethylene from alcohol and cocaine. This ability to catalyze a transesterification is also reflected by flumazenil esterase which forms in vitro flumazenil methylester from flumazenil acid and methanol but not from flumazenil acid in the presence of methanol (Figure 1). This result indicates a further similarity of both proteins and may raise speculations about a potential interference of the flumazenil and cocaine degradation pathway. In different cellular fractions the activity of the formation of flumazenil acid and flumazenil methylester was predominantly found in the microsomes but in addition low amounts of flumazenil methylester activity was detected in the mitochondrial and cytosolic fraction without observation of flumazenil acid formation. This differential cellular distribution of both activities can be attributed to another unknown enzyme catalyzing only the transesterification reaction. Further investigations using cytosol or mitochondria as biological source for a protein isolation procedure in connection with the transesterification assay will allow to distinguish this enzyme from the presented carboxylesterase from human liver microsomes. The formation of N-demethylated flumazenil is probably catalyzed by cytochrome P-450. The metabolite was detected only in the presence of a NADPH-generating system and the activity of the responsible P-450 enzyme depended on the chosen solvent. In the presence of acetonitrile and acetone the highest activities were found. It is well known that cytochrome P-450 1A2, 2C19, 2D6 and 3A isozymes are involved in N-dealkylation reactions in human liver microsomes (). For example the N-demethylation of cocaine in rat liver microsomes is catalyzed by cytochrome P-450 2B1 (30). Because of the small amounts of N-demethylated flumazenil which are produced by incubation of human liver microsomes it is not possible to determine the responsible P-450 subfamily by simple inhibition experiments. Additional investigations using recombinantly expressed P-450 isozymes are in progress to further characterize the cytochrome P-450 enzyme subfamily catalyzing the N-demethylation of flumazenil. AcknowledgementsThe authors wish to thank Mrs. B. Endreß and Mrs. P. Zaremba for the excellent technical assistance and Mr F. Distler from the Institute of Forensic Medicine, University of Erlangen-Nürnberg, for performing the GC-MS analyses. ReferencesMöhler H; Richard JG. Agonist and antagonist benzodiazepine receptor interaction in vitro. Nature 294:763-765, 1981. Amrein R; Hetzel W. Pharmacology of Dormicum midazolam and Anexate flumazenil. Acta Anaesthesiol Sc. 34:Suppl.92: 6-15, 1990. Brouard A; Perdu E; Bismuth C. RO 15-1788 Monograph. Journal de Toxicologic Clinique et Expérimentale T.7, N°4:223-237, 1987. Klotz U; Kanto J. Pharmacokinetics and clinical use of flumazenil Ro 15-1788. Clin. Pharmacokinet. 14:1-12, 1988. Vletter AA; Burm AGL; Breimer LTM; Spierdijk J. High-performance liquid chromatographic assay to determine midazolam and flumazenil simultaneously in human plasma. Biomedical Applications 177-185, 1990. Hunkeler W. Preclinical research findings with flumazenil Ro 15-1788, Anexate: chemistry. Eur. J. Anaesthesiol. suppl 2:37-62, 1988. Kronbach T; Mathys D; Umeno M; Gonzalez FJ ; Meyer UA: Oxidation of midazolam and triazolam by human liver cytochrome P450 IIIA4. Mol. Pharmacol. 36:89-96, 1989. Andersson T; Miners JO; Veronese ME; Birkett DJ: Diazepam metabolism by human liver microsomes is mediated by both S-mephenytoin hydrolase ; CYP3A isoforms. Br. J. clin. Pharmacol . 38:131-137 1994. Guengerich FP. Oxidative Cleavage of Carboxylic Esters by Cytochrome P-450. J. Biol. Chem. 263:8176-8183, 1988. Kleingeist B; Böcker R; Geisslinger G; Brugger R. In vitro metabolism of flumazenil by a human liver carboxyl esterase and cyctochrome P450. Exp. Toxic. Pathol. Suppl II 48:194-201, 1996. Kleingeist B, Böcker R, Brugger R. Purification and characterization of a microsomal human liver flumazenil carboxylesterase. Naunyn Schmiedeberg´s Arch. Pharmacol., 353, Suppl., R 112. Brugger R; García Alía B; Reichel C; Waibel R; Menzel S; Brune K; Geisslinger G. Isolation and characterization of rat liver microsomal R-ibuprofenoyl-CoA synthetase. Biochem. Pharmacol. 52:1007-1013, 1996. Bradford MM. A rapid and sensitive method for quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248-254, 1976. Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685, 1970. Wray W; Boulikas T; Wray VP; Hancock R. Silver staining of proteins in polyacrylamide gels. Anal. Biochem. 118:197-203, 1981. Hofstee BHJ. On the evaluation of the constants Vmax and KM in enzyme reactions. Science 116:329-331, 1952. Harano T; Miyata T; Lee S; Aoyagi H; Omura T. Biosynthesis and localization of rat liver microsomal carboxylesterase E1. J. Biochem. Tokyo 103:149-155, 1988. Mentlein R; Shumann M; Heymann E. Comparative chemical and immunological characterization of five lipolytic enzmes carboxylesterases from rat liver microsomes. Arch. Biochem. Biophys. 234:612-621, 1984. Heymann E; Junge W. Characterization of the isoenzymes of pig-liver esterase. 1. Chemical studies. Eur. J. Biochem. 95:509-518, 1979. Long RM; Calabrese MR; Martin BM; Pohl LR. Cloning and sequencing of a human liver carboxylesterase isoenzyme. Life Sci. 48:PL43-PL49, 1991. Munger JS; Shi G-P; Mark EA; Chin DT; Gerard C; Chapman HA. A serine esterase released by human alveolar macrophages is closely related to liver microsomal carboxylesterases. J Biol Chem 266:18832-18838 1991. Jakoby WB. Detoxication enzymes, in Jakoby WB (ed): Enzymatic Basis of Detoxification, Vol. I. Academic Press, New York, pp 1-6, 1980. Heymann E; Junge W. Characterization of the isoenzymes of pig-liver esterase. 1. Chemical studies. Eur. J. Biochem. 95:509-518, 1979. Ketterman AJ; Bowles MR; Pond SM. Purification and characterization of two human liver carboxylesterases. Int. J. Biochem. 21:1303-1312, 1989. Robbi M; Beaufay H. Purification and characterization of various esterases from rat liver. Eur. J. Biochem. 137:293-301, 1983. Walker CH. Esterases: Problems of identification and classification. Biochem. Pharmacol. 32:3265-3269, 1986. Brzezinski MR; Abraham TL; Stone CL; Dean RA; Bosron WF. Purification and characterization of a human liver cocaine carboxylesterase that catalyzes the production of benzoylecgonine and the formation of cocaethylene from alcohol and cocaine. Biochem. Pharmacol. 48:1747-1755, 1994. Coutts RT; Su P; Baker GB. Involvement of CYP 2D6, CYP 3A4, and other cytochrome P-450 isozymes in N-dealkylation reactions. JPM 31:177-186, 1994. Footnotes 1 Present address, F. Hoffmann-La Roche Ltd., Vitamins and Fine Chemicals Division, Department of Biotechnology, CH-4070 Basel, Switzerland. Corresponding author: Gerd Geisslinger Department of Experimental and Clinical Pharmacology and Toxicology, University of Erlangen-Nürnberg, Universitätsstr. 22, D-91054 Erlangen, Germany.
Published by the Canadian Society for Pharmaceutical Sciences. Copyright © 1997 by the Canadian Society for Pharmaceutical Sciences. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||