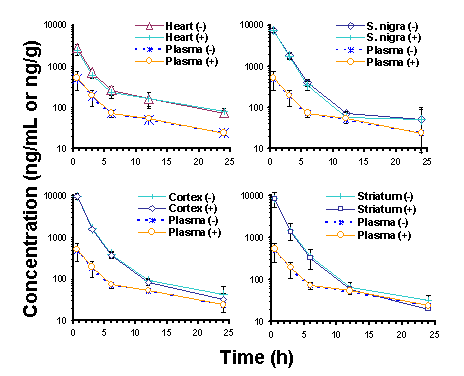
Disposition of ethopropazine enantiomers in the rat: Tissue distribution and plasma protein binding. Manuscript received April 24th, 1999; Revised April 28th, 1999, Accepted April 29th, 1999. Mojdeh Maboudian-Esfahani Dion R. Brocks1 Presented in part at the 1998 American Association of Pharmaceutical Sciences Annual Meeting, San Francisco, CA, USA Abstract Purpose. To determine the in vitro plasma protein binding, and the in vivo brain, heart and plasma concentrations of ethopropazine (ET) enantiomers in the rat after iv doses. Methods. For in vivo assessment of ET enantiomer concentrations, rats with implanted jugular vein cannulae were injected with 10 mg/kg of (± )-ET HCl. At selected times after dosing, rats were sacrificed and heart, brain, and plasma were collected. Equilibrium dialysis was used to determine the unbound fraction of ET in rat plasma over a concentration range of 150 to 4000 ng/mL of each enantiomer. A stereospecific assay was used to measure concentrations of ET enantiomer. Results. No stereoselectivity was observed in plasma or tissues after iv dosing. Area under the concentration vs. time curves indicated that highest uptake of ET occurred in brain tissue, followed by heart tissues, then plasma. There was no noticeable difference between concentrations of ET enantiomers in different parts of brain (substantia nigra, cortex, or striatum). There was no observed stereoselectivity in plasma protein binding of ET enantiomers in rat plasma. Saturation of binding to plasma proteins was observed between 500 and 2000 ng/mL of each ET enantiomer, but unbound fraction was constant at concentrations below and above that range. Conclusion: Ethopropazine displays nonstereoselectivity in its pharmacokinetics. The drug shares distribution features similar to those of other phenothiazine derivatives. Based on the in vitro plasma protein binding results, there appears to be saturation of some, but not all, plasma binding proteins of ET within the range of concentrations studied. Introduction Ethopropazine is a phenothiazine compound used for its anticholinergic properties in the treatment of Parkinson's disease. Although the drug has been in clinical use for over 30 y, there is no information available regarding its pharmacokinetic properties in humans. In the rat, ET exhibits a large volume of distribution, and in comparison to hepatic blood flow, it is relatively slowly cleared from plasma (1). The drug has a long terminal phase elimination half-life of >20 h, and negligible amounts of unchanged drug are recoverable in urine and bile after iv doses (1). Upon increasing the dose of ET HCl from 5 to 10 mg/kg, there is a less than proportionate increase in AUC, suggesting that a nonlinear process is involved in its disposition. After oral doses are administered, the drug displays very poor systemic bioavailability of < 5% (1). Similar to other anticholinergic drugs used in the treatment of Parkinson's disease, ET is chiral and is used clinically as the racemate. This may be relevant in viewing ET pharmacokinetics since stereoselectivity in the binding of other chiral anticholinergic drugs, including procyclidine and trihexyphenidyl, to muscarinic receptors has been demonstrated (2). Muscarinic receptors are present in many tissues in the body, and there are several subtypes that are classified on the basis of their anatomical distribution. There is no information available regarding the stereoselective properties of ET enantiomers. Despite the previous study (1)), there were still some issues related to the pharmacokinetics of ET in the rat that were unresolved. Given that ET is chiral and is administered as the racemate, the possibility of stereoselectivity in its pharmacokinetics was of interest. In addition, there was no information related to tissue uptake of the drug or plasma protein binding of ET enantiomers. In this study the tissue and plasma concentrations of its ET enantiomers in the rat after intravenous dosing are reported. Heart and brain tissues were studied owing to their importance as target tissues of anticholinergic drugs from the perspective of therapeutics and toxicity. Plasma protein binding of ET enantiomers was also examined. Methods and Materials Animals Under halothane anesthesia, a Silastic cannula was inserted into the right jugular vein of male Sprague-Dawley rats (mean wt = 314 g). The rats were then allowed an overnight rest. The next day, ET was dissolved in a stock dosing solution containing ethanol:polypropylene glycol:water (10:30:60 v/v). After diluting the stock solution 5-fold in 5% dextrose in water for injection, the drug was administered intravenously (10 mg/kg) via the jugular vein cannula. At 0.5, 3, 6, 12 and 24 h after dosing, the rats (3 rats per time) were anesthetized with halothane and decapitated. Blood was collected and centrifuged at 2500 g for 10 min to permit separation of plasma. Plasma, brain and heart tissues were stored at -20ºC until assayed for ET enantiomers. On the day of analysis the tissue samples were thawed, and three segments of brain (cortex, substantia nigra, striatum) were identified and dissected with the aid of a microscope. Plasma protein binding studies For the study of binding of ET enantiomers to plasma proteins, the pH of blank rat plasma was measured and adjusted to pH 7.4 by addition of 0.5 M HCl (~7 m L acid/mL of plasma). After addition of appropriate amounts of (± )-ET stock solution to clean test tubes, the solution was dried under nitrogen. Then, 1 mL of blank rat plasma was added, and the tubes were vortex mixed for 2 min. In vitro plasma protein binding was determined by equilibrium dialysis using a Spectrum dialysis apparatus (Los Angeles, CA, USA) and Sigma Diagnostics dialysis sacks (St. Louis, MO, USA) (molecular weight cut off = 12,000). The dialysis membrane was initially boiled for 5 min in double distilled water. After two sequential rinses in double distilled water, the sacks were placed in isotonic pH 7.4 Sorensens phosphate buffer for 10 min before being cut and fitted between 1 mL Teflon dialysis cells. One half of each dialysis cell was filled with drug-free 0.9 ml isotonic Sorensen’s phosphate buffer (pH = 7.4), and the other side was filled with an equal volume of plasma spiked with racemic ET (300, 1000, 4000 and 8000 ng/mL). Each concentration was run in quadruplicate. The cells were placed into the Spectrum apparatus, then dialyzed in a water bath at 37° C under constant rotation for 3 h. To calculate the extent of volume shift during dialysis, the concentration of protein in the plasma was compared before and after dialysis using a modification of the Folin-Lowry method (Bio-Rad DC Protein Assay Kit, Hercules, CA. USA). For analysis of ET in the dialyzed samples, 100 and 700 m L aliquots of plasma and buffer, respectively, were measured and the concentrations of ET enantiomers were determined using stereospecific HPLC. Assay For assay of ET enantiomers in rat plasma, a previously developed stereospecific high performance liquid chromatographic method was utilized (3). For analysis of enantiomer concentrations in brain and heart tissues, some modification of the assay was required. Tissue samples (~20 mg) were weighed and placed in glass mortar and pestle tissue grinders. A volume of 0.3 mL sodium phosphate buffer (pH 5.9) was added, along with 60 m L of 13 m g/mL ( ± )-diphenidol HCl in methanol. The tissues were homogenized using hand-driven tissue grinders, and the samples were transferred to new polypropylene centrifuge tubes. Proteins were precipitated by addition of 0.3 mL acetonitrile while the tubes were vortex mixed. The tubes were subsequently centrifuged at 2000 g for 4 min and the supernatants were carefully transferred to new glass tubes using Pasteur pipets. From this step the samples were extracted with hexane in the same manner as deproteinized plasma samples (3). Standard curves were prepared for each tissue type using spiked tissue homogenates. Quality control samples were incorporated into each analytical run to assure integrity of the results. Data Analysis In order to estimate the plasma concentration immediately after dosing, compartmental models were fitted to the mean plasma concentration vs. time data using SAAM II (SAAM Institute, Seattle, WA USA). The area under the plasma and tissue concentration vs. time curves (AUC) from the time of dosing to the last measured concentration was estimated using the combined log-linear trapezoidal rule. The unbound fraction in plasma (fu) was determined by: Statistical analysis of the pharmacokinetic data was performed using the Student’s paired or unpaired t-test, as appropriate. A p value of less than 0.05 was considered statistically significant. ANOVA was used to assess the significance of the differences in binding of ET to plasma proteins at different concentrations. Duncan’s Multiple range test was used to rank the plasma protein binding of ET enantiomers at different concentrations. All data were expressed as a mean ± SD, unless otherwise stated. Results Modification of the assay resulted in linear standard curves (r2>0.99) in each of the tissues studied, with concentrations ranging from 125 to 2500 ng/mL. Quality control data of concentrations of 125 and 1250 ng/g of ET enantiomer in tissues indicated that the mean relative SD and bias were <7%. After iv dosing, the enantiomer concentrations were almost superimposable over the first 24 hours after dosing in plasma and each of the tissues (Figure 1). Although there were quantitative differences between the plasma and tissue enantiomer concentration vs. time curves, they each displayed qualitative similarities in their patterns of decline (Figure 1). The highest concentrations observed in each of the tissues were attained at 0.5 h after dosing (Figure 1). There were no noticeable differences in ET enantiomer concentrations between nigrostriatum, striatum, and cortex (Figure 1, Table 1). The relative AUC0-24h indicated that concentrations in brain tissues were higher than those in heart, followed by plasma (Table 1). The tissue:plasma AUC ratios were between 6 and 8 in brain segments compared to approximately 3.5 in heart. The uptake of ET in each of the three brain segments was of the same order of magnitude.
The suitability of both ultrafiltration and equilibrium dialysis for the measurement of the degree of plasma protein binding of ET was assessed. Ultrafiltration was first tried using Centrifree micropartition systems (Amicon, Danvers, MA, USA). It was found that the recovery of ET was low due to extensive (>84%) binding of drug to the filter of the Centrifree system. This prevented the use of ultrafiltration to determine the unbound fraction of ET in plasma. Equilibrium dialysis was then attempted to determine the plasma protein binding of ET enantiomers. Based on assay of the plasma and buffer content after dialysis, and comparing the results to the assay of the plasma and buffer contents of ET before dialysis, it was concluded that there was no binding of ET to the dialysis membrane. Therefore equilibrium dialysis could be used to assess the plasma protein binding of ET.
There was no difference in protein concentration before and after dialysis, indicating that a significant volume shift did not occur during dialysis. In a preliminary study, there were no differences in the fraction of ET bound to plasma proteins when dialysis was run for 2 hours as compared to 4 hours. Hence, dialysis was run for 3 hours in the protein binding experiments. There was no binding of ET enantiomers to the dialysis membranes. Ethopropazine enantiomers were extensively bound to plasma proteins (>95%) over a wide range of concentrations (Table 2). There was no significant difference in unbound fraction between plasma ET enantiomer concentrations of 150 and 500 ng/mL. Analysis using Duncan's Multiple range test, however, indicated that the unbound fractions at these lower concentrations were significantly lower than those at the higher plasma enantiomer concentrations of 2000 and 4000 ng/mL. The unbound fraction at 4000 ng/mL was not significantly different from that at 2000 ng/mL. Discussion The pharmacokinetics of ET enantiomers has been studied previously in a single rat (3). No stereoselectivity in plasma concentrations was observed in that animal. This study confirms the lack of stereoselectivity in ET pharmacokinetics; at no time point, in plasma or tissues, did the (+):(-) ratio of ET enantiomers significantly deviate from unity in any of the specimens from any of the animals studied (Table 1, Figure 1). Further, there were no significant differences noted in the binding of the enantiomers to plasma proteins (Table 2).
Since tissue distribution can be a major determinant of drug effect, it is important to understand the relationship between concentration of drug in plasma and the sites where pharmacological receptors are located; e.g., muscarinic receptors for anticholinergics (2). Consequently, the concentrations of ET in plasma were compared to those in localized segments of brain and heart, tissues in which muscarinic receptors are present. There were notable differences between the AUC of ET enantiomers in brain, heart and plasma. The mean Vd of (± )-ET has been reported to be greater than 12 L/kg (1). The greater concentrations of ET enantiomers in the tissues as compared to plasma are consistent with the large Vd. In each of the examined anatomical regions of brain, concentrations were similar and higher than those in heart and plasma. Ethopropazine is a lipophilic drug with relatively low aqueous solubility. Brain tissue contains abundant quantities of lipids, including sphingomyelin and phosphatidyl choline, which would be expected to promote uptake of a lipophilic drug such as ET (4). Recently it has been suggested that intralysosomal sequestration is partially responsible for high tissue concentrations of the anticholinergic drugs used in Parkinson's disease (5). The distribution of ET enantiomers to brain and heart tissues was rapid, Cmax being attained at or before 0.5 h after administration of drug (Figure 1). The pattern of distribution of ET in brain was very similar to that reported for chlorpromazine in rat brain (6). Relative to the AUC0-24 in plasma, chlorpromazine accumulated 21 times more in brain and 11 times more in heart. In our study, the AUC0-24 of ET in brain and heart were ~7-fold and 4-fold higher than in plasma, respectively. The uptake of the phenothiazine derivatives chlorpromazine, promethazine, perphenazine and triflupromazine were studied in different parts of murine brain (7). One h after iv dosing, the concentrations of each drug in brain were similar and about 40-fold higher than in blood. However, similar to ET, the drug concentrations were not significantly different between different parts of the brain. As indicated in Table 2, it was estimated that the unbound fraction of ET enantiomers in plasma was less than 5.5%. This higher extent of binding is shared with other phenothiazine derivatives, including chlorpromazine, trifluoperazine, thioridazine and perazine (8-10). An important observation was that there appeared to be saturable binding of ET enantiomers to rat plasma over the range of ET concentrations used in our study. The percentage of unbound ET enantiomers remained constant over the concentration range of 150 to 500 ng/mL (Table 2). However, between 500 to 2000 ng/mL there was a significant increase in the unbound fraction of ET enantiomers. Moreover, with an increase in ET enantiomer concentration from 2000 to 4000 ng/mL, there was no significant difference in free fraction (Table 2). This finding is consistent with binding of ET enantiomers to more than one plasma protein, and has been reported previously for other phenothiazines (8). Upon increasing the concentration of phenothiazines from 100 to 400 ng/mL, no saturation of binding to isolated human serum albumin, lipoprotein or g -globulin proteins occurs (8). On the other hand, binding of phenothiazines to a 1-acid glycoprotein over the same concentration range was saturated. Also, the binding of chlorpromazine to plasma proteins was decreased with concentrations >400 ng/mL. Each of the plasma proteins studied (8) was involved in the binding of these basic drugs. At least one of the proteins involved in the binding of ET appears to be saturated over the range of concentrations studied in the present study (Table 2), whereas binding to other proteins is not. The saturable protein likely possesses a high degree of affinity, but low capacity, for ET (e.g., a 1-acid glycoprotein). The non-saturable protein would likely have low affinity but high capacity for binding of the drug (e.g., albumin). After iv doses of 5 and 10 mg/kg (± )-ET HCl given to rats, the increase in AUC was less than proportional in relation to the dose (1). This was accompanied by a significant increase in CL and Vdb at the 10 mg/kg dose level, and suggested that the pharmacokinetics of ET were nonlinear between 5 and 10 mg/kg. These results were in line with those of the plasma protein binding observed in the present study, where nonlinearity was apparent in the binding of ET enantiomers to plasma proteins between 500 and 2000 ng/mL (Table 2). To date the pharmacokinetics of ET has only been reported in the rat. Whether these findings have relevance in humans has yet to be established. Usually Vd of drugs is similar between species, and one would anticipate that there is also a large Vd in humans. Other findings in the rat, such as CL, pathways of elimination, and stereoselectivity, can only be put into context in the face of actual data from human subjects. Acknowledgements This work was funded by a grant from the Health Services Utilization Commission of Saskatchewan. The authors are grateful to Dr. Adil Nazarali of the University of Saskatchewan for his helpful discussions and guidance in performing the brain dissections. References
Corresponding author: Dion R. Brocks, College of Pharmacy, Western University of Health Sciences, 309 E. 2nd St., College Plaza, Pomona, CA, USA. dbrocks@western.edu Keywords: pharmacokinetics, anticholinergics, Parkinson's disease Published by the Canadian Society for Pharmaceutical Sciences. Copyright © 1999 by the Canadian Society for Pharmaceutical Sciences. |
|||||||||||||||||||||||||||||||||||||||||||||||||||